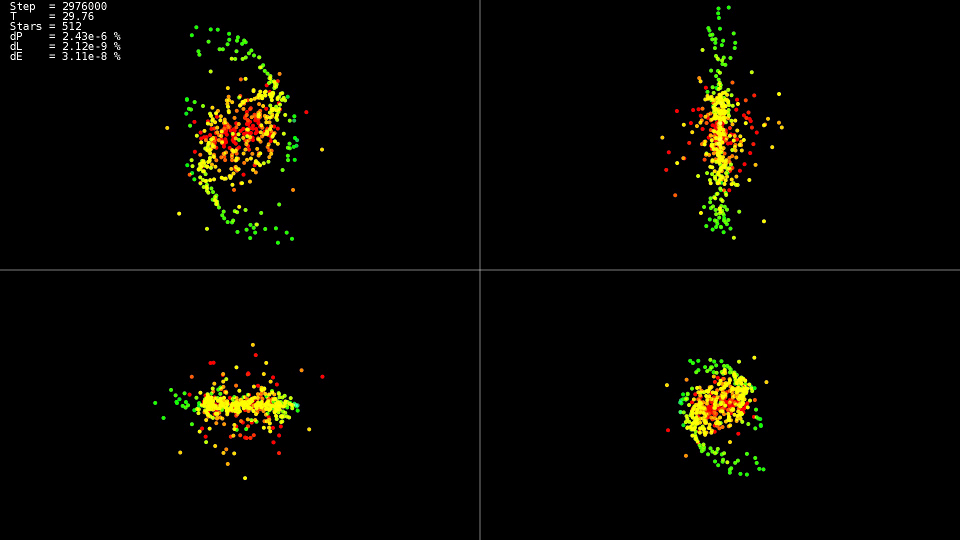
No hace mucho leí la novela de ciencia ficción
"El problema de los tres cuerpos" de Liu Qixin . En él, algunos extraterrestres tenían un problema: no sabían cómo, con suficiente precisión para ellos, calcular la trayectoria de su planeta de origen. A diferencia de nosotros, vivían en un sistema de tres estrellas, y el "clima" del planeta dependía en gran medida de su disposición mutua, desde el calor incinerador hasta el frío helado. Y decidí verificar si podemos resolver tales problemas.
Física del fenómeno.
Para comprender el problema, es necesario tratar con la física del fenómeno. En el marco de la teoría clásica, la fuerza de atracción de dos cuerpos está determinada por
la ley
de Newton :
v e c F ( v e c r 1 , v e c r 2 ) = - G m 1 m 2 f r a c v e c r 1 - v e c r 2 | v e c r 1 - v e c r 2 | 3 ,
donde
vecr1, vecr2 - la posición de los cuerpos en el espacio,
m1,m2 - masas de cuerpos,
G - constante gravitacional.
En el sistema de
N Los cuerpos en cada uno de ellos se verán afectados por la fuerza de atracción del resto, que se expresa mediante la ecuación:
vecFn=−G sumk neqnmnmk frac vecrn− vecrk| vecrn− vecrk|3.$
Usando
la segunda ley de Newton, escribimos la aceleración para cada partícula:
vecan= vecFn/mn=−G sumk neqnmk frac vecrn− vecrk| vecrn− vecrk|3.$
Recordando que la aceleración es la segunda derivada del tiempo de la coordenada, obtenemos una ecuación diferencial parcial de segundo orden, que debe resolverse para obtener la trayectoria de cada cuerpo:
frac partial2 vecrn partialt2=fn=−G sumk neqnmk frac vecrn− vecrk| vecrn− vecrk|3.$
Es importante señalar aquí que la complejidad de calcular una función
fn es igual a
O(N2) y aumenta significativamente con el aumento del número de cuerpos que interactúan.
Matemáticas
El primer y más simple método para resolver ecuaciones diferenciales es
el método de Euler , que está diseñado para resolver ecuaciones de la forma:
fracdydx=f(x,y).$
Tras la transición a la región discreta, obtenemos:
yi=yi−1+hf(xi−1,yi−1), quadi=1,2,3, puntos,m,
donde
h Es el paso de integración, y
m - el número de pasos de integración. Por lo tanto, si necesitamos calcular la posición de los cuerpos a la vez
T entonces deberíamos hacer
m=T/h pasos de integración. Aquí el primer problema es visible, si
T grande, entonces debemos tomar una gran cantidad de pasos de integración.
Para aplicar el método de Euler a nuestro problema, debe reducirse a un sistema de primer orden. Para hacer esto, presentamos una variable adicional: velocidad de partículas:
frac partial vecvn partialt=fn, frac partial vecrn partialt= vecvn.$
El segundo problema para resolver sistemas de ecuaciones diferenciales es la precisión de la solución y su control. La precisión se puede mejorar de dos maneras: disminuyendo el paso de integración y eligiendo un método con un mayor orden de precisión. Ambos métodos conducen a una mayor complejidad computacional, pero de diferentes maneras. Por ejemplo, puede usar el
método clásico
Runge-Kutta de cuarto orden ; requiere cuatro cálculos de funciones
fn en cada paso, pero tiene un orden de precisión
O(h4) (a modo de comparación, el método de Euler tiene un orden de precisión
O(h) y requiere un cálculo
fn ) La precisión de la solución también se puede controlar de varias maneras: compare con la solución analítica, resuelva utilizando diferentes métodos o con diferentes pasos y compare los resultados, controle los parámetros de terceros y las limitaciones que la solución debe cumplir.
Además, cada uno de estos métodos tiene sus inconvenientes. Las decisiones analíticas pueden estar ausentes o, incluso en la mayoría de los casos, completamente ausentes. Por ejemplo, para nuestra tarea
N la solución analítica de tel es solo para
N=2 , pero incluso esto es suficiente para probar la precisión de los métodos. Resolver un problema mediante dos métodos o con diferentes pasos aumenta el tiempo de cálculo, pero este enfoque se puede aplicar a casi cualquier tarea. No todas las tareas tienen limitaciones, pero las nuestras las tienen: en cada paso de integración podemos controlar la implementación
de las leyes de conservación . Este enfoque también aumenta la complejidad del cálculo, pero hay mucho para elegir; calcular la suma de los momentos momentáneos o angulares de todas las partículas tiene una complejidad del orden
O(N) , mientras que calcular la energía total de un sistema tiene complejidad de orden
O(N2)Nota sobre el cálculo de la energía totalEn nuestro caso, la energía total del sistema consta de dos partes: energía cinética y energía potencial. La energía cinética consiste en la suma de las energías cinéticas de todos los cuerpos. Para calcular la energía potencial, necesitamos agregar las energías potenciales de cada partícula en el campo gravitacional de las partículas restantes, por lo que debemos agregar
O(N2) términos La dificultad es que todos los términos son de un orden muy diferente, e incluso con cálculos de doble precisión no es posible calcular este valor con una precisión suficiente para comparar en diferentes pasos. Para superar este problema, fue necesario aplicar la suma de acuerdo con
el algoritmo de Kahan .
Fig 1. Un ejemplo de una trayectoria elíptica.
Considere el caso simple de un satélite que se mueve en una órbita elíptica alrededor de la Tierra. A medida que el satélite se acerca a la Tierra, su velocidad aumenta, y al alejarse de la Tierra disminuye, en consecuencia, surge la posibilidad de disminuir el paso de integración en el tiempo en la parte verde de la órbita, y aumentarlo en rojo y azul sin cambiar la precisión de la solución. Intentemos comparar con más detalle.
Tabla 1. Métodos investigados para resolver ecuaciones diferencialesPara seleccionar el mejor método para nuestra tarea, compararemos varios métodos conocidos. Para hacer esto, simulamos la colisión de dos sistemas de cuerpos.
N=512 y mida el cambio relativo en la
energía total , el
momento y su
momento al final de la simulación (tiempo máximo de simulación)
Tmax=2.5 ) En este caso, variaremos el paso y los parámetros de los métodos de integración y mediremos el número de llamadas a funciones
fn , respectivamente, aquellos métodos que con un menor número de llamadas conducirán a una menor pérdida, consideraremos más aceptables.

| 
|
| a) | b) |
| Figura 2. Cambio relativo en energía a), momento b), al final de la simulación del sistema N=512 cuerpos por varios métodos dependiendo del número de cálculos de funciones fn doble precisión |
De los gráficos de la Figura 2 se puede ver que la mejor relación entre la cantidad de cálculo de la función
fn y el cambio relativo en la energía del sistema de cuerpos en los métodos Adams y Dorman-Prince de quinto orden. También se ve que para todos los métodos con un aumento en el número de cálculos
fn El cambio relativo en el impulso del sistema aumenta. Para un cambio relativo en la energía, esto también es notable, pero solo para algunos métodos que podrían alcanzar el umbral
dE/E0<10−12 . Este efecto ya no se puede atribuir al error de los métodos, sino al error de los cálculos, y un aumento adicional en la precisión solo es posible junto con un aumento en la precisión de los cálculos con coma flotante.

| 
|
| a) | b) |
| Fig. 3. Cambio relativo en energía a), momento b), al final de la simulación del sistema N=512 cuerpos por varios métodos dependiendo del número de cálculos de funciones fn con precisión cuádruple (__float128) |
Las Figuras 3a y 3b muestran que el uso de cálculos con precisión cuádruple puede reducir las pérdidas de energía relativas hasta
10−23 , pero debe comprender que el tiempo de cálculo aumenta en dos órdenes de magnitud en comparación con la precisión doble. Como en el caso de los cálculos de doble precisión, la mejor relación de precisión y el número de cálculos de funciones
fn poseen métodos de quinto orden Adams y Dorman-Prince.
Los
métodos Dorman-Prince y Werner pertenecen a la clase de
métodos anidados y pueden calcular simultáneamente dos soluciones con un orden de precisión alto y bajo (para el método Dorman-Prince, órdenes 5 y 4, y para el método Werner, órdenes 6 y 5). Si estas dos soluciones son muy diferentes, entonces podemos dividir el paso de integración actual en otras más pequeñas. Eso nos permite cambiar dinámicamente el paso de integración y reducirlo solo en aquellas áreas donde se requiere.
Comparemos los métodos de quinto orden de Dorman-Prince, Werner y Adams con más detalle, durante un intervalo de simulación más largo de nuestro sistema (
Tmax=300 )

|
a) El cambio relativo en energía, momento y su momento en el proceso de modelado
|

|
b) Número de cálculos de funciones fn en el intervalo de simulación DeltaT=0.3
|
Fig. 4. Dependencias del cambio relativo en los parámetros físicos del sistema y el número de cálculos de funciones. fn de vez en cuando modelando con el método de paso variable Dorman-Prince h=10−5 puntos10−9
|

|
a) El cambio relativo en energía, momento y su momento en el proceso de modelado
|

|
b) Número de cálculos de funciones fn en el intervalo de simulación DeltaT=0.3
|
Fig. 5. Dependencias del cambio relativo en los parámetros físicos del sistema y el número de cálculos de funciones. fn versus modelado de tiempo por el método de Werner con un paso variable h=10−5 puntos10−9
|

|
Fig. 6. Cambio relativo en energía, momento y su momento en el proceso de modelado por el método Adams-Bashfort de quinto orden con un paso h=10−6
|

|
Fig 7. Dependencias del número de cálculos de funciones. fn para métodos Adams, Dorman-Prince y Werner de quinto orden a partir del tiempo de simulación
|
Se puede ver que en la etapa inicial de la evolución de nuestro sistema (
T<25 ) los tres métodos muestran características similares, pero en etapas posteriores se producen algunos eventos en el sistema, como resultado de los cuales los errores en los parámetros principales del sistema (energía total, momento y su momento) saltan bruscamente. Pero los métodos de Dorman-Prince y Werner hacen frente a estos cambios mucho mejor debido a la capacidad de reducir el paso de integración en secciones "complejas", como resultado de lo cual aumenta el número de cálculos de funciones
fn como se ve en las Figuras 4b y 5b, pero el número total de cálculos
fn Los métodos anidados son más pequeños que el método Adams-Bashfort, como se puede ver en la Figura 7.
Me pregunto qué pasó con el sistema en estos momentos.
|
Video 1. Modelado de un sistema de 512 cuerpos. Método Dorman-Prince. Paso dinámico h=10−5 puntos10−9
|
El video demuestra eso hasta el momento
T=25 el movimiento es relativamente tranquilo, y después de una colisión de los centros de las "galaxias", lo que conduce a un cambio brusco en las trayectorias y un fuerte aumento en las velocidades de algunas partículas. Además, para mantener la precisión de la solución, es necesario reducir el paso de integración. Los métodos anidados pueden hacer esto automáticamente; los gráficos muestran que en algunas partes de la evolución del sistema, el paso de integración se redujo en casi dos órdenes de magnitud con
10−5 antes
h=10−7 . Al usar el método Adams y tal paso en todo el intervalo de la evolución del sistema, no hubiéramos esperado una solución.
Resumen
Para la solución, es mejor utilizar métodos anidados que le permitan controlar dinámicamente el paso de integración y reducirlo solo en secciones "complejas" de la trayectoria.
No persiga los métodos de orden superior. Incluso cuando usan el tipo de datos 'doble', no alcanzan sus capacidades potenciales, el uso de los tipos de datos con mayor precisión aumenta en gran medida el tiempo que lleva resolver el problema.
Implementación de la CPU
Ahora que la elección de un método para resolver ecuaciones está definida, intentemos calcular el cálculo de la fuerza de interacción para cada partícula. Obtenemos un doble ciclo para todas las partículas:
Código de implementación 'simple'for(size_t body1 = 0; body1 < count; ++body1) { const nbvertex_t v1(rx[ body1 ], ry[ body1 ], rz[ body1 ]); nbvertex_t total_force; for(size_t body2 = 0; body2 != count; ++body2) { if(body1 == body2) { continue; } const nbvertex_t v2(rx[ body2 ], ry[ body2 ], rz[ body2 ]); const nbvertex_t force(m_data->force(v1, v2, mass[body1], mass[body2])); total_force += force; } frx[body1] = vx[body1]; fry[body1] = vy[body1]; frz[body1] = vz[body1]; fvx[body1] = total_force.x / mass[body1]; fvy[body1] = total_force.y / mass[body1]; fvz[body1] = total_force.z / mass[body1]; }
Las fuerzas gravitacionales para cada cuerpo se calculan de forma independiente, y para usar todos los núcleos del procesador, es suficiente escribir la directiva OpenMP antes del primer ciclo:
Un fragmento de código de la implementación de 'openmp' #pragma omp parallel for for(size_t body1 = 0; body1 < count; ++body1)
Porque Dado que cada cuerpo interactúa con cada uno, para reducir la cantidad de interacciones del procesador con la RAM y mejorar el uso de la memoria caché, tenemos la capacidad de cargar parte de los datos en la memoria caché y usarla repetidamente:
Código de implementación 'openmp + block' #pragma omp parallel for for(size_t n1 = 0; n1 < count; n1 += BLOCK_SIZE) { nbcoord_t x1[BLOCK_SIZE]; nbcoord_t y1[BLOCK_SIZE]; nbcoord_t z1[BLOCK_SIZE]; nbcoord_t m1[BLOCK_SIZE]; nbvertex_t total_force[BLOCK_SIZE]; for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; x1[b1] = rx[local_n1]; y1[b1] = ry[local_n1]; z1[b1] = rz[local_n1]; m1[b1] = mass[local_n1]; } for(size_t n2 = 0; n2 < count; n2 += BLOCK_SIZE) { nbcoord_t x2[BLOCK_SIZE]; nbcoord_t y2[BLOCK_SIZE]; nbcoord_t z2[BLOCK_SIZE]; nbcoord_t m2[BLOCK_SIZE]; for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { size_t local_n2 = b2 + n2; x2[b2] = rx[local_n2]; y2[b2] = ry[local_n2]; z2[b2] = rz[local_n2]; m2[b2] = mass[n2 + b2]; } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { const nbvertex_t v1(x1[ b1 ], y1[ b1 ], z1[ b1 ]); for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { const nbvertex_t v2(x2[ b2 ], y2[ b2 ], z2[ b2 ]); const nbvertex_t force(m_data->force(v1, v2, m1[b1], m2[b2])); total_force[b1] += force; } } } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; frx[local_n1] = vx[local_n1]; fry[local_n1] = vy[local_n1]; frz[local_n1] = vz[local_n1]; fvx[local_n1] = total_force[b1].x / m1[b1]; fvy[local_n1] = total_force[b1].y / m1[b1]; fvz[local_n1] = total_force[b1].z / m1[b1]; } }
Una optimización adicional consiste en extraer el contenido de la función de calcular la fuerza en el ciclo principal y eliminar la división y multiplicación por la masa corporal m1 [b1]. Además del hecho de que hemos reducido un poco los cálculos, el compilador podrá aplicar instrucciones vectoriales del procesador SSE y AVX en un ciclo tan extendido.
Código de implementación 'openmp + block + optimization' #pragma omp parallel for for(size_t n1 = 0; n1 < count; n1 += BLOCK_SIZE) { nbcoord_t x1[BLOCK_SIZE]; nbcoord_t y1[BLOCK_SIZE]; nbcoord_t z1[BLOCK_SIZE]; nbcoord_t total_force_x[BLOCK_SIZE]; nbcoord_t total_force_y[BLOCK_SIZE]; nbcoord_t total_force_z[BLOCK_SIZE]; for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; x1[b1] = rx[local_n1]; y1[b1] = ry[local_n1]; z1[b1] = rz[local_n1]; total_force_x[b1] = 0; total_force_y[b1] = 0; total_force_z[b1] = 0; } for(size_t n2 = 0; n2 < count; n2 += BLOCK_SIZE) { nbcoord_t x2[BLOCK_SIZE]; nbcoord_t y2[BLOCK_SIZE]; nbcoord_t z2[BLOCK_SIZE]; nbcoord_t m2[BLOCK_SIZE]; for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { size_t local_n2 = b2 + n2; x2[b2] = rx[local_n2]; y2[b2] = ry[local_n2]; z2[b2] = rz[local_n2]; m2[b2] = mass[n2 + b2]; } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { nbcoord_t dx = x1[b1] - x2[b2]; nbcoord_t dy = y1[b1] - y2[b2]; nbcoord_t dz = z1[b1] - z2[b2]; nbcoord_t r2(dx * dx + dy * dy + dz * dz); if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = (m2[b2]) / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; total_force_x[b1] -= dx; total_force_y[b1] -= dy; total_force_z[b1] -= dz; } } } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; frx[local_n1] = vx[local_n1]; fry[local_n1] = vy[local_n1]; frz[local_n1] = vz[local_n1]; fvx[local_n1] = total_force_x[b1]; fvy[local_n1] = total_force_y[b1]; fvz[local_n1] = total_force_z[b1]; } }
Tabla 2. Dependencia del tiempo de cálculo (en segundos) de la función fn sobre la cantidad de cuerpos que interactúan N para diversas implementaciones de CPU| N | 2048 | 4096 | 8192 | 16384 | 32768 |
|---|
| simple | 0,0425 | 0.1651 | 0.6594 | 2,65 | 10,52 |
| Openmp | 0.0078 | 0,0260 | 0,1079 | 0.417 | 1.655 |
| openmp + block + optimización | 0.0037 | 0,0128 | 0,0495 | 0,194 | 0,774 |
Parámetros del sistema:- sistema: Debian 9, Intel Core i7-5820K (6 núcleos)
- compilador: gcc 6.3.0
Se ve claramente que la versión con soporte de OpenMP se acelera seis veces, exactamente en términos de la cantidad de núcleos, y la versión optimizada es más rápida un poco más de dos veces. Entonces, con la optimización, no debe confiar solo en la concurrencia. Curiosamente, en los cálculos de flujo único, el procesador trabajó a una frecuencia de 3.6 GHz, en la versión paralela (openmp) bajó la frecuencia a 3.4 GHz, y en el paralelo y optimizado (openmp + block + optimización) bajó a 3.3 GHz, pero esto no le impedía trabajar 13,6 veces más rápido. También se ve que un aumento en el tiempo de cálculo con un aumento en el tamaño del problema es cuadrático, y un aumento adicional
N hace que la tarea sea insoluble en un tiempo razonable.
Implementación de GPU
Pero existe el deseo de hacer cálculos aún más rápidos. Hay varias direcciones disponibles para la aceleración: cálculo de GPU, aproximación de funciones
fn . Primero, para la informática de GPU, elegí la tecnología OpenCL. Para un trabajo más conveniente, se
utilizó la biblioteca
CLHPP . La principal ventaja de OpenCL es que el código se puede ejecutar en el procesador y en la GPU, lo que simplifica la escritura y la depuración, además de expandir la lista de hardware para ejecutar. La herramienta
Oclgrind ayuda en la depuración, que en tiempo de ejecución muestra llamadas incorrectas a la API OpenCL y problemas de acceso a la memoria.
Opencl
Para comenzar con OpenCL, debe obtener una lista de las plataformas disponibles. Las plataformas más comunes son AMD, Intel y NVidia.
Código std::vector<cl::Platform> platforms; cl::Platform::get(&platforms);
Luego, después de elegir una plataforma, debe seleccionar el dispositivo informático que representa esta plataforma:
Código const cl::Platform& platform(platforms[platform_n]); std::vector<cl::Device> all_devices; platform.getDevices(CL_DEVICE_TYPE_ALL, &all_devices);
Y al final de la fase preparatoria, es necesario crear un contexto y colas dentro de las cuales se asignará la memoria y se realizarán los cálculos. Por ejemplo, un contexto que combina todos los dispositivos informáticos de una plataforma seleccionada se crea de la siguiente manera:
Contexto y código de creación de cola cl::Context context(all_devices); std::vector<cl::CommandQueue> queues; for(cl::Device device: all_devices) queues.push_back(cl::CommandQueue(context, device));
Para descargar el código fuente a un dispositivo informático, debe compilarse, la clase cl :: Program está destinada a esto.
Código de compilación del núcleo std::vector< std::string > source_data; cl::Program::Sources sources; for(int i = 0; i != files.size(); ++i) { source_data.push_back(load_program(files[i]));
Para describir los parámetros de una función (kernel) que se ejecuta en un dispositivo informático, hay una plantilla cl: make_kernel.
Ejemplo de núcleo de cálculo de fuerza de interacción typedef cl::make_kernel< cl_int, cl_int,
Además, todo es simple: declaramos una variable con el tipo de núcleo, pasamos el programa compilado y el nombre del núcleo computacional, podemos iniciar el núcleo casi como una función normal.
Código de lanzamiento del kernel ComputeBlock fcompute(prog, "ComputeBlockLocal"); cl::NDRange global_range(device_data_size); cl::NDRange local_range(block_size); cl::EnqueueArgs eargs(ctx.m_queue, global_range, local_range); fcompute(eargs, ... );
El núcleo computacional para OpenCL en sí es muy similar a la opción 'openmp + block + optimization' para la CPU, solo que a diferencia de la versión de la CPU, el primer ciclo se controla usando OpenCL (el rango del ciclo está determinado por la variable global_range del código de inicio del kernel, y el número de iteración actual está disponible desde el kernel utilizando la función get_global_id (0)). Primero, parte de los datos del cuerpo se cargan desde la memoria local y luego se procesan. La memoria local es común a todos los subprocesos del grupo, por lo que la descarga se produce una vez para el grupo y cada subproceso del grupo la procesa y, dado que la memoria local es mucho más rápida que la global, los cálculos son mucho más rápidos.
Código del núcleo __kernel void ComputeBlockLocal(int offset_n1, int offset_n2, __global const nbcoord_t* mass, __global const nbcoord_t* y, __global nbcoord_t* f, int yoff, int foff, int points_count, int stride) { int n1 = get_global_id(0) + offset_n1; __global const nbcoord_t* rx = y + yoff; __global const nbcoord_t* ry = rx + stride; __global const nbcoord_t* rz = rx + 2 * stride; __global const nbcoord_t* vx = rx + 3 * stride; __global const nbcoord_t* vy = rx + 4 * stride; __global const nbcoord_t* vz = rx + 5 * stride; __global nbcoord_t* frx = f + foff; __global nbcoord_t* fry = frx + stride; __global nbcoord_t* frz = frx + 2 * stride; __global nbcoord_t* fvx = frx + 3 * stride; __global nbcoord_t* fvy = frx + 4 * stride; __global nbcoord_t* fvz = frx + 5 * stride; nbcoord_t x1 = rx[n1]; nbcoord_t y1 = ry[n1]; nbcoord_t z1 = rz[n1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; __local nbcoord_t x2[NBODY_DATA_BLOCK_SIZE]; __local nbcoord_t y2[NBODY_DATA_BLOCK_SIZE]; __local nbcoord_t z2[NBODY_DATA_BLOCK_SIZE]; __local nbcoord_t m2[NBODY_DATA_BLOCK_SIZE];
Cuda
La implementación de la plataforma NVidia CUDA es un poco más simple que OpenCL, no necesitamos crear el contexto del dispositivo y administrar la cola de ejecución nosotros mismos (al menos hasta que queramos hacer una implementación multi-GPU). Como en el caso de OpenCL, necesitamos asignar memoria en la GPU, copiar nuestros datos en ella y luego podemos iniciar el núcleo informático.
Puede leer más sobre trabajar con CUDA
aquí .
Código de lanzamiento del kernel de CUDA dim3 grid(count / block_size); dim3 block(block_size); size_t shared_size(4 * sizeof(nbcoord_t) * block_size); kfcompute <<< grid, block, shared_size >>> (... ...);
A diferencia de OpenCL, CUDA no especifica el rango completo de iteraciones (en la implementación de OpenCL es global_range), pero establece el tamaño de cuadrícula y los tamaños de bloque en la cuadrícula, en consecuencia, el cálculo del número de cuerpo actual cambia ligeramente, de lo contrario, el núcleo es muy similar a OpenCL, con la excepción de otros nombres funciones de sincronización y especificación para memoria compartida. Otra característica distintiva útil de CUDA es que podemos especificar el tamaño requerido de memoria compartida cuando se inicia el núcleo. Al igual que en la implementación de OpenCL, al comienzo de cada bloque de iteración copiamos parte de los datos en la memoria compartida y luego trabajamos con esta memoria desde todos los hilos del bloque.
Código del núcleo CUDA __global__ void kfcompute(int offset_n2, const nbcoord_t* y, int yoff, nbcoord_t* f, int foff, const nbcoord_t* mass, int points_count, int stride) { int n1 = blockDim.x * blockIdx.x + threadIdx.x; const nbcoord_t* rx = y + yoff; const nbcoord_t* ry = rx + stride; const nbcoord_t* rz = rx + 2 * stride; nbcoord_t x1 = rx[n1]; nbcoord_t y1 = ry[n1]; nbcoord_t z1 = rz[n1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; extern __shared__ nbcoord_t shared_xyzm_buf[]; nbcoord_t* x2 = shared_xyzm_buf; nbcoord_t* y2 = x2 + blockDim.x; nbcoord_t* z2 = y2 + blockDim.x; nbcoord_t* m2 = z2 + blockDim.x; for(int b2 = 0; b2 < points_count; b2 += blockDim.x) { int n2 = b2 + offset_n2 + threadIdx.x;
Tabla 3. La dependencia de la función de tiempo de cálculo (en segundos) fn sobre la cantidad de cuerpos que interactúan N para diversas implementaciones de GPU y mejores implementaciones de CPU| N | 4096 | 8192 | 16384 | 32768 | 65536 | 131072 |
|---|
| openmp + block + optimización | 0,0128 | 0,0495 | 0,194 | 0,774 | --- | --- |
| OpenCL + half NVidia K80 | 0.004 | 0.008 | 0,026 | 0,134 | 0.322 | 1,18 |
| CUDA + half NVidia K80 | 0.004 | 0.008 | 0,0245 | 0,115 | 0.291 | 1.13 |
Donde conseguir NVidia K80Las implementaciones de OpenCL y CUDA se lanzaron en el servicio gratuito
Colab de Google, que proporciona acceso a las computadoras NVidia K80. Puede leer más sobre cómo trabajar con este servicio en el
concentrador En general, el resultado es decepcionante, solo 5-6 veces más rápido que la implementación de la CPU. Incluso si realizamos cálculos en todo el K80, obtendremos una aceleración de hasta 12 veces, pero desde Dado que la complejidad de la tarea es cuadrática, en un tiempo razonable podremos procesar no 32768 cuerpos interactuando, sino 131072, que es solo 4 veces más.
Aproximación de funciones fn
Si observa de cerca la función, que se establece por la fuerza de atracción de dos cuerpos, está claro que disminuye cuadráticamente con la distancia. Por lo tanto, podemos calcular con precisión la fuerza de interacción entre cuerpos cercanos, y aproximadamente entre cuerpos distantes. Un enfoque famoso
es un algoritmo de
código de árbol propuesto por D. Barnes y P. Hat. Se construye un
árbol de octree en el espacio simulado, que contiene en sus hojas las coordenadas y masas de los cuerpos simulados. Los nodos principales contienen el centro de masa, la masa total de los nodos secundarios y el radio de la esfera descrita alrededor de los cuerpos de los nodos secundarios. La raíz del árbol contiene el centro de masa de todos los cuerpos, su masa total y el radio de la esfera que se describe a su alrededor. Al calcular la fuerza de interacción, la distancia a la raíz del árbol se considera primero si la relación de la distancia al nodo a su radio es mayor que alguna constante
lambdacrit , entonces la raíz se considera un cuerpo con coordenadas iguales a las coordenadas del centro de masa de los cuerpos incluidos en él, y una masa igual a la suma de las masas de los nodos hijos, si la relación es menor o igual a
lambdacrit , el procedimiento se repite recursivamente para cada nodo secundario. Si se alcanza la hoja de un árbol, la fuerza de interacción se considera de la manera habitual. Por lo tanto, si un cuerpo está muy alejado del grupo compacto de otros cuerpos, este grupo se le presenta como un cuerpo, y la fuerza de interacción se calcula no hasta cada cuerpo, sino solo hasta un cuerpo. Debido a esto, la complejidad del algoritmo disminuye con
O(N2) antes
O(N log(N)) a costa de alguna pérdida de precisión.
En mi enfoque, decidí no usar el
árbol de octree , sino el
árbol de kd, porque es más fácil de usar y tiene menos gastos de almacenamiento en comparación con el octree.
Volvamos a la implementación en la CPU. Un nodo de árbol kd se puede representar en forma de una clase que contiene punteros a los descendientes izquierdo y derecho e información sobre las coordenadas y la masa:
Nodo de árbol Kd class node { node* m_left;
Con este método de almacenamiento del árbol, tenemos dos opciones posibles para atravesar el árbol: usar la recursividad explícita o usar la pila nosotros mismos. Me decidí por la segunda opción.
Cálculo de la fuerza de interacción atravesando un árbol nbvertex_t force_compute(const nbvertex_t& v1, const nbcoord_t mass1) { nbvertex_t total_force; node* stack_data[MAX_STACK_SIZE] = {}; node** stack = stack_data; node** stack_head = stack; *stack++ = m_root; while(stack != stack_head) { node* curr = *--stack; const nbcoord_t distance_sqr((v1 - curr->m_mass_center).norm()); if(distance_sqr > curr->m_radius_sqr) {
Como en el caso de la implementación de CPU "exacta", se llama a la función de cálculo de fuerza para cada cuerpo. El ciclo en todos los cuerpos se puede paralelizar fácilmente con las directivas OpenMP.
Pero las iteraciones de bucle vecino en este caso se referirán a partes completamente diferentes del árbol, lo que no permite el uso eficiente de la memoria caché del procesador. Para superar este problema, todos los cuerpos deben atravesarse no en el orden original, sino en el orden en que se ubican en las hojas del árbol kd, entonces se producirán iteraciones vecinas para los cuerpos que están cerca del espacio, y atravesarán el árbol a lo largo de casi los mismos caminos.
Transversal de la hoja del árbol template<class Visitor> void traverse(Visitor visit) { node* stack_data[MAX_STACK_SIZE] = {}; node** stack = stack_data; node** stack_head = stack; *stack++ = m_root; while(stack != stack_head) { node* curr = *--stack; if(curr->m_radius_sqr > 0) {
Esta implementación tiene otro problema: no hay una forma universal de paralelizar tal recorrido de árbol. Pero podemos cambiar completamente la forma en que se almacena el árbol en la memoria: podemos almacenar todos los nodos en una matriz lineal y abandonar por completo el almacenamiento de punteros a los descendientes, por analogía con la construcción de un
montón binario . Al comenzar la numeración de nodos con
1 si el nodo está en el número de celda
i , entonces su hijo izquierdo está en la celda
2i , niño derecho en la celda
2i+1 y el padre en la celda
i/2 . El nodo derecho correspondiente a la izquierda con el número
i tendrá un número
i+1 . La matriz en sí tendrá una longitud
2N , y todos los nodos hoja se ubicarán en el último
N Además, las celdas, los nodos muy espaciados se ubicarán en celdas cercanas de la matriz. La función transversal del árbol cambiará un poco, pero el marco sigue siendo el mismo:
Cálculo de la fuerza atravesando un árbol en una matriz nbvertex_t force_compute(const nbvertex_t& v1, const nbcoord_t mass1) { nbvertex_t total_force; size_t stack_data[MAX_STACK_SIZE] = {}; size_t* stack = stack_data; size_t* stack_head = stack; *stack++ = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { size_t curr = *--stack; const nbcoord_t distance_sqr((v1 - m_mass_center[curr]).norm()); if(distance_sqr > m_radius_sqr[curr]) { total_force += force(v1, m_mass_center[curr], mass1, m_mass[curr]); } else { size_t left(left_idx(curr)); size_t rght(rght_idx(curr)); if(rght < m_body_n.size()) { *stack++ = rght; } if(left < m_body_n.size()) { *stack++ = left; } } } return total_force; }
Pero estas no son todas las posibilidades que nos abre el almacenamiento de los nodos en la matriz: podemos rechazar la pila al rastrear. Para hacer esto, en la rama de código en la que vamos a los hijos del nodo, agregamos la función para calcular el siguiente nodo (
nextup ), y en la rama en la que calculamos la fuerza de interacción, agregamos el cálculo del siguiente nodo con el subárbol actual omitido (
s k i p d o w n )
Para omitir el subárbol actual, debemos bajar a la raíz (dirección
D ), mientras estamos en el descendiente derecho, tan pronto como lleguemos a la izquierda, vamos al subárbol derecho correspondiente (dirección
R ), si llegamos a la raíz, se completa el recorrido del árbol.
Código de función de omisión de subárbol index_t skip_down(index_t idx) {
 Figura 8. Saltarse un subárbol s k i p d o w n .
Figura 8. Saltarse un subárbol s k i p d o w n .Para ir al siguiente nodo, si es posible, vaya al niño izquierdo (dirección
U ), y si no hay descendiente, vaya al siguiente nodo 'desde abajo' utilizando la función
s k i p d o w n .
Ir al siguiente código de función de nodo index_t next_up(index_t idx, index_t tree_size) { index_t left = left_idx(idx); if(left < tree_size) { return left; } return skip_down(idx); }
 Figura 9. Transiciones al siguiente nodo n e x t u p .
Figura 9. Transiciones al siguiente nodo n e x t u p .Puede parecer que reemplazamos la recursión con un bucle
w h i l e en función
s k i p d o w n , y dicho reemplazo no da nada, pero veamos cómo determinar si un nodo con un número
yo descendiente derecho Esto se puede hacer simplemente comprobando su número impar (el elemento secundario correcto está en la celda
2 i + 1 ), para esto es suficiente calcular
i \ & 1 . Es decir dividimos el número
i dos si el bit menos significativo se establece en uno. Pero esto se puede hacer sin un bucle, en muchos procesadores hay una instrucción
find first set que devuelve la posición del primer bit set, usándolo convertimos el bucle en cuatro instrucciones de procesador.
Código de función de subárbol omitido optimizado index_t skip_down(index_t idx) { idx = idx >> (__builtin_ffs(~idx) - 1); return left2right(idx); }
Después de eso, podemos excluir la pila de la función transversal del árbol y reemplazarla con un par
skipdown+nextup , después de eso la función se ve aún más simple:
Cálculo de la fuerza atravesando un árbol en una matriz sin usar una pila nbvertex_t force_compute(const nbvertex_t& v1, const nbcoord_t mass1) const { nbvertex_t total_force; size_t curr = NBODY_HEAP_ROOT_INDEX; size_t tree_size = m_mass_center.size(); do { const nbcoord_t distance_sqr((v1 - m_mass_center[curr]).norm()); if(distance_sqr > m_radius_sqr[curr]) { total_force += force(v1, m_mass_center[curr], mass1, m_mass[curr]); curr = skip_down(curr); } else { curr = next_up(curr, tree_size); } } while(curr != NBODY_HEAP_ROOT_INDEX); return total_force; }
En total, obtuvimos seis combinaciones de recorrido de árbol y cálculo de fuerza. Compare estos enfoques en términos de tiempo de cálculo y calidad. Tomamos como medida de calidad el cambio relativo en la energía total del sistema después de 100 iteraciones. Como sistema modelo, tomamos dos "galaxias" interactivas que consisten en16384cuerpos cada uno.Tabla 4. Combinaciones de método de recorrido de árbol y cálculo de fuerza| Tipo de cálculo transversal del árbol / fuerza | Árbol con pila | 'Pila' con una pila | 'Montón' sin pila |
|---|
| Iteraciones por número de cuerpo | ciclo + árbol | ciclo + montón | ciclo + heapstackless |
|---|
| Desvío de hojas | árbol + árbol anidado | nestedtree + heap | nestedtree + heapstackless |
|---|

| 
|
| a) Dependencia del tiempo de cálculo de la función fn desde la relación de la distancia crítica al nodo del árbol a su radio ( λcrit ) | b) La dependencia del cambio relativo en energía en el sistema en la relación de la distancia crítica al nodo del árbol a su radio ( λcrit ) |
| Figura 10. Resultados del cálculo de funciones fn de varias maneras en la CPU (número de cuerpos N=32768 ) |
Se puede ver que la implementación de 'árbol anidado + árbol' está irremediablemente atrasado en velocidad, porque Carece de concurrencia. Las implementaciones con la ubicación de los nodos de árbol en la matriz y la indexación como en un montón binario están a la cabeza. El cambio relativo en la energía es insignificante para todas las variantes conλcrit>1 .
También en la fig. 10a muestra que para (λcrit<10 ) todas las variantes del cálculo de funciones fn adelanta significativamente en velocidad la versión más optimizada del cálculo exacto ('openmp + block + optimization'), con un aumento adicional λcrit Las implementaciones con un árbol pierden la versión exacta.Recorrido de árbol de GPU
Traté de evitar el árbol en la GPU usando la tecnología OpenCL y CUDA. La opción de almacenar nodos en forma de árbol se descartó de inmediato, y solo quedaban las opciones para almacenar el árbol en una matriz con indexación como en un montón binario. En general, la implementación del núcleo informático no es muy diferente de la versión de la CPU.Kernel OpenCL para calcular la fuerza al atravesar un árbol (transversal en el orden de los cuerpos de numeración) __kernel void ComputeTreeBH(int offset_n1, int points_count, int tree_size, __global const nbcoord_t* y, __global nbcoord_t* f, __global const nbcoord_t* tree_cmx, __global const nbcoord_t* tree_cmy, __global const nbcoord_t* tree_cmz, __global const nbcoord_t* tree_mass, __global const nbcoord_t* tree_crit_r2) { int n1 = get_global_id(0) + offset_n1; int stride = points_count; __global const nbcoord_t* rx = y; __global const nbcoord_t* ry = rx + stride; __global const nbcoord_t* rz = rx + 2 * stride; __global const nbcoord_t* vx = rx + 3 * stride; __global const nbcoord_t* vy = rx + 4 * stride; __global const nbcoord_t* vz = rx + 5 * stride; __global nbcoord_t* frx = f; __global nbcoord_t* fry = frx + stride; __global nbcoord_t* frz = frx + 2 * stride; __global nbcoord_t* fvx = frx + 3 * stride; __global nbcoord_t* fvy = frx + 4 * stride; __global nbcoord_t* fvz = frx + 5 * stride; nbcoord_t x1 = rx[n1]; nbcoord_t y1 = ry[n1]; nbcoord_t z1 = rz[n1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int stack_data[MAX_STACK_SIZE] = {}; int stack = 0; int stack_head = stack; stack_data[stack++] = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { int curr = stack_data[--stack]; nbcoord_t dx = x1 - tree_cmx[curr]; nbcoord_t dy = y1 - tree_cmy[curr]; nbcoord_t dz = z1 - tree_cmz[curr]; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > tree_crit_r2[curr]) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tree_mass[curr] / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; } else { int left = left_idx(curr); int rght = rght_idx(curr); if(left < tree_size) { stack_data[stack++] = left; } if(rght < tree_size) { stack_data[stack++] = rght; } } } frx[n1] = vx[n1]; fry[n1] = vy[n1]; frz[n1] = vz[n1]; fvx[n1] = res_x; fvy[n1] = res_y; fvz[n1] = res_z; }
En la primera versión, el recorrido del árbol comenzó en el orden de numeración de los cuerpos en la matriz original, por lo que los subprocesos vecinos omitieron partes completamente diferentes del árbol, lo que afectó negativamente el rendimiento de la memoria caché de la GPU. Por lo tanto, en la segunda realización, se aplicó un recorrido, comenzando desde la parte superior del árbol, en este caso, los hilos vecinos comienzan a atravesar el árbol a lo largo del mismo camino, porque las copas de los árboles vecinos están cerca y en el espacio. También es importante que elijamos la numeración en el conjunto de nodos del árbol no desde cero, sino desde uno, en este caso las hojas del árbol se almacenan en la segunda mitad del conjunto, y con el número de cuerpos igual a la potencia de dos, tendremos el mismo acceso a la memoria por el índice tn1.Kernel OpenCL para calcular la fuerza atravesando un árbol (atravesando los nodos de numeración de un árbol) __kernel void ComputeHeapBH(int offset_n1, int points_count, int tree_size, __global const nbcoord_t* y, __global nbcoord_t* f, __global const nbcoord_t* tree_cmx, __global const nbcoord_t* tree_cmy, __global const nbcoord_t* tree_cmz, __global const nbcoord_t* tree_mass, __global const nbcoord_t* tree_crit_r2, __global const int* body_n) { int tree_offset = points_count - 1 + NBODY_HEAP_ROOT_INDEX; int stride = points_count; int tn1 = get_global_id(0) + offset_n1 + tree_offset; int n1 = body_n[tn1]; nbcoord_t x1 = tree_cmx[tn1]; nbcoord_t y1 = tree_cmy[tn1]; nbcoord_t z1 = tree_cmz[tn1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int stack_data[MAX_STACK_SIZE] = {}; int stack = 0; int stack_head = stack; stack_data[stack++] = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { int curr = stack_data[--stack]; nbcoord_t dx = x1 - tree_cmx[curr]; nbcoord_t dy = y1 - tree_cmy[curr]; nbcoord_t dz = z1 - tree_cmz[curr]; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > tree_crit_r2[curr]) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tree_mass[curr] / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; } else { int left = left_idx(curr); int rght = rght_idx(curr); if(left < tree_size) { stack_data[stack++] = left; } if(rght < tree_size) { stack_data[stack++] = rght; } } } __global const nbcoord_t* vx = y + 3 * stride; __global const nbcoord_t* vy = y + 4 * stride; __global const nbcoord_t* vz = y + 5 * stride; __global nbcoord_t* frx = f; __global nbcoord_t* fry = frx + stride; __global nbcoord_t* frz = frx + 2 * stride; __global nbcoord_t* fvx = frx + 3 * stride; __global nbcoord_t* fvy = frx + 4 * stride; __global nbcoord_t* fvz = frx + 5 * stride; frx[n1] = vx[n1]; fry[n1] = vy[n1]; frz[n1] = vz[n1]; fvx[n1] = res_x; fvy[n1] = res_y; fvz[n1] = res_z; }
Al recorrer el orden de numeración de los nodos del árbol, obtuvimos un aumento en el rendimiento. Pero esta opción también se puede mejorar. La memoria global en la que se encuentran actualmente los nodos del árbol está optimizada para acceso compartido , es decir Los hilos de un grupo deben leer palabras ubicadas en un bloque de memoria. En nuestro caso, el recorrido del árbol comienza a lo largo de los mismos caminos, y solicitamos los mismos datos con todos los hilos del grupo, pero cuanto más profundizamos en el árbol, los caminos de los hilos vecinos divergen cada vez más, y tenemos que solicitar datos diferentes, lo que reduce el rendimiento del subsistema varias veces memoria Pero los nodos de cada subárbol que pertenecen al mismo nivel se encuentran en celdas de memoria relativamente cercanas. Es decir
Al atravesar el resto del árbol, los subprocesos vecinos del núcleo informático no acceden a los mismos nodos del árbol, sino que se encuentran en la memoria. Para optimizar este acceso a la memoria, se puede utilizar la memoria de textura. Pero hay un inconveniente. Por el momento, las texturas no admiten trabajar con datos de doble precisión (queremos calcular con precisión). Pero en CUDA hay una función __hiloint2double que recopila un número de doble precisión de dos enteros.Código de solicitud de número de precisión doble de una textura que almacena enteros template<> struct nb1Dfetch<double> { typedef double4 vec4; static __device__ double fetch(cudaTextureObject_t tex, int i) { int2 p(tex1Dfetch<int2>(tex, i)); return __hiloint2double(py, px); } static __device__ vec4 fetch4(cudaTextureObject_t tex, int i) { int ii(2 * i); int4 p1(tex1Dfetch<int4>(tex, ii)); int4 p2(tex1Dfetch<int4>(tex, ii + 1)); vec4 d4 = {__hiloint2double(p1.y, p1.x), __hiloint2double(p1.w, p1.z), __hiloint2double(p2.y, p2.x), __hiloint2double(p2.w, p2.z) }; return d4; } };
En este caso, se realizaron dos implementaciones, en una cada elemento del árbol (x, y, z, tree_crit_r2) se solicitó de forma independiente, y en la segunda implementación se combinaron estas solicitudes. La solicitud de la masa del nodo se produce con mucha menos frecuencia, solo si se cumple la condición r2> tree_crit_r2 [curr] , por lo que no tiene sentido combinar esta solicitud con las demás. Otra característica útil del marco CUDA es la capacidad de controlar la proporción de los tamaños del caché L1 y el tamaño de la memoria compartida ( cudaFuncSetCacheConfig ). En el caso del recorrido del árbol, no usamos memoria compartida, por lo que podemos aumentar el caché L1 en detrimento de él.Kernel CUDA para calcular la fuerza atravesando un árbol (atravesando el orden de numeración de los nodos del árbol) __global__ void kfcompute_heap_bh_tex(int offset_n1, int points_count, int tree_size, nbcoord_t* f, cudaTextureObject_t tree_xyzr, cudaTextureObject_t tree_mass, const int* body_n) { nb1Dfetch<nbcoord_t> tex; int tree_offset = points_count - 1 + NBODY_HEAP_ROOT_INDEX; int stride = points_count; int tn1 = blockDim.x * blockIdx.x + threadIdx.x + offset_n1 + tree_offset; int n1 = body_n[tn1]; nbvec4_t xyzr = tex.fetch4(tree_xyzr, tn1); nbcoord_t x1 = xyzr.x; nbcoord_t y1 = xyzr.y; nbcoord_t z1 = xyzr.z; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int stack_data[MAX_STACK_SIZE] = {}; int stack = 0; int stack_head = stack; stack_data[stack++] = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { int curr = stack_data[--stack]; nbvec4_t xyzr2 = tex.fetch4(tree_xyzr, curr); nbcoord_t dx = x1 - xyzr2.x; nbcoord_t dy = y1 - xyzr2.y; nbcoord_t dz = z1 - xyzr2.z; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > xyzr2.w) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tex.fetch(tree_mass, curr) / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; } else { int left = nbody_heap_func<int>::left_idx(curr); int rght = nbody_heap_func<int>::rght_idx(curr); if(left < tree_size) { stack_data[stack++] = left; } if(rght < tree_size) { stack_data[stack++] = rght; } } } f[n1 + 3 * stride] = res_x; f[n1 + 4 * stride] = res_y; f[n1 + 5 * stride] = res_z; }
Un análisis del programa en el perfilador nvprof mostró que incluso cuando se usa memoria de textura para almacenar un árbol, todavía hay una carga muy alta en la memoria global.De hecho, en CUDA, toda la memoria del núcleo que se dirige a direcciones 'calculadas' se almacena en la memoria global y, en consecuencia, la pila que se necesita para atravesar el árbol se encuentra en la memoria global y consume una parte significativa del ancho de banda del chip de memoria, porque la pila tiene cada hilo de ejecución, y hay muchos hilos.Pero, afortunadamente, ya sabemos cómo atravesar un árbol sin usar una pila. Complementando el núcleo computacional anterior con las funciones de calcular el siguiente nodo de árbol, obtenemos un nuevo núcleo, además, más compacto.Núcleo CUDA para la fuerza informática al atravesar un árbol sin usar una pila __global__ void kfcompute_heap_bh_stackless(int offset_n1, int points_count, int tree_size, nbcoord_t* f, cudaTextureObject_t tree_xyzr, cudaTextureObject_t tree_mass, const int* body_n) { nb1Dfetch<nbcoord_t> tex; int tree_offset = points_count - 1 + NBODY_HEAP_ROOT_INDEX; int stride = points_count; int tn1 = blockDim.x * blockIdx.x + threadIdx.x + offset_n1 + tree_offset; int n1 = body_n[tn1]; nbvec4_t xyzr = tex.fetch4(tree_xyzr, tn1); nbcoord_t x1 = xyzr.x; nbcoord_t y1 = xyzr.y; nbcoord_t z1 = xyzr.z; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int curr = NBODY_HEAP_ROOT_INDEX; do { nbvec4_t xyzr2 = tex.fetch4(tree_xyzr, curr); nbcoord_t dx = x1 - xyzr2.x; nbcoord_t dy = y1 - xyzr2.y; nbcoord_t dz = z1 - xyzr2.z; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > xyzr2.w) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tex.fetch(tree_mass, curr) / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; curr = nbody_heap_func<int>::skip_idx(curr); } else { curr = nbody_heap_func<int>::next_up(curr, tree_size); } } while(curr != NBODY_HEAP_ROOT_INDEX); f[n1 + 3 * stride] = res_x; f[n1 + 4 * stride] = res_y; f[n1 + 5 * stride] = res_z; }
El rendimiento de los núcleos que se ejecutan en la GPU depende en gran medida del tamaño de los bloques en los que dividimos la tarea. Dependiendo de este tamaño, cuántos registros, memoria local y otros recursos estarán disponibles para cada hilo informático. También debe tener en cuenta que mientras espera el acceso a la memoria en un subproceso, otro subproceso puede realizar cálculos en el procesador del sombreador, por lo tanto, con un número suficiente de subprocesos ejecutados simultáneamente en un procesador, el tiempo de acceso a la memoria estará oculto detrás de los cálculos. Por lo tanto, antes de comparar el rendimiento de nuestros núcleos, necesitamos calcular el tamaño de bloque óptimo para cada uno de ellos. Hagamos una comparación de la mitad disponible de NVidia K80.Tabla 5. La dependencia de la función de tiempo de cálculo (en segundos) fn GPU N=131072 λcrit=10| / | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 1024 |
|---|
| opencl+dense | 5.77 | 2.84 | 1.46 | 1.13 | 1.15 | 1.14 | 1.14 | 1.13 |
| cuda+dense | 5.44 | 2.55 | 1.27 | 0.96 | 0.97 | 0.99 | 0.99 | - |
| opencl+heap+cycle | 5.88 | 5.65 | 5.74 | 5.96 | 5.37 | 5.38 | 5.35 | 5.38 |
| opencl+heap+nested | 4.54 | 3.68 | 3.98 | 5.25 | 5.46 | 5.41 | 5.48 | 5.31 |
| cuda+heap+nested | 3,62 | 2,81 | 2.68 | 4.26 | 4.84 | 4.75 | 4.8 | 4.67 |
| cuda+heap+nested+tex | 2.6 | 1.51 | 0.912 | 0.7 | 1.85 | 1.75 | 1.69 | 1.61 |
| cuda+heap+nested+tex+stackless | 2.3 | 1.29 | 0.773 | 0.5 | 0.51 | 0.52 | 0.52 | 0.52 |
6. ( ) fn GPU N=1M λ c r i t = 4| Tamaño de bloque / núcleo | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 1024 |
|---|
| opencl + denso | 366 | 179 | 89,9 | 69,3 | 70,3 | 69,1 | 68,9 | 68,0 |
| cuda + denso | 346 | 162 | 79,6 | 60,8 | 60,8 | 60,7 | 59,6 | - |
| opencl + montón + ciclo | 16,2 | 18,2 | 20,1 | 21,2 | 21,2 | 21,3 | 21,2 | 21,1 |
| opencl + montón + anidado | 10,5 | 7.63 | 6.38 | 8.23 | 9,95 | 9,89 | 9.65 | 9.66 |
| cuda + montón + anidado | 8.67 | 6.44 | 5.39 | 5.93 | 8.65 | 8.61 | 8.41 | 8.27 |
| cuda + montón + anidado + tex | 6.38 | 3,57 | 2,13 | 1,44 | 3,56 | 3.46 | 3.30 | 3.29 |
| cuda + montón + anidado + tex + sin pila | 5,78 | 3.19 | 1,83 | 1,21 | 1.11 | 1.10 | 1.11 | 1.13 |
Una situación difícil, pero, a diferencia de la versión de CPU del recorrido del árbol, está claro que cada paso de optimización trae resultados tangibles. La implementación de 'opencl + heap + cycle' es casi 6 veces más lenta que una solución exacta con cálculo de función completaf n .
Una implementación de 'opencl + heap + nested', en la que un recorrido del árbol comienza desde los nodos vecinos, 1.4 veces más rápido que el anterior, porque La memoria caché se usa mejor. En la implementación de 'cuda + heap + nested', el caché L1 se aumentó en detrimento de la memoria compartida, lo que aumentó la velocidad en 1,4 veces, aunque es posible que en la implementación de cuda el núcleo de compilación se compile de manera más óptima (en las versiones de 'opencl + dense' y 'cuda Los núcleos + densos son idénticos, y el rendimiento de la versión de Cuda es ~ 1.2 veces mayor). El aumento más significativo en la velocidad computacional (3.8 veces) se logra cuando el árbol se encuentra en la memoria de textura y se combinan las consultas a los elementos del nodo del árbol. La implementación con el recorrido del árbol sin usar la pila 'cuda + heap + nested + tex + stackless' es 1.4 veces más rápida también.en él, todo el ancho de banda del bus de memoria se usa solo para acceder a datos sobre nodos de árbol y no se gasta en la pila. Así, conλ c r i t = 10 fue posible lograr la aceleración dos veces en comparación con el cálculo completo de la funciónf n . Pero
λ c r i t = 10 es un valor excesivamente grande del parámetro; en el gráfico del cambio relativo de energía en el sistema versus la relación de la distancia crítica al nodo del árbol a su radio para la implementación de la CPU, está claro que los valores más bajosλ c r i t sin aparente pérdida de precisión de la solución. Intentemos variarλ c r i t en los tamaños de bloque óptimos, que determinamos en el paso anterior.
| 
|
a) N = 128 K
| b) N = 1 M
|
Fig. 11. Dependencia del tiempo de cálculo de la función. f n desde la relación de la distancia crítica al nodo del árbol a su radio ( λ c r i t ) para varias implementaciones de GPU Tree Walk
|
Se puede ver que para pequeños λ c r i t todos los métodos de cálculo de la funciónf n para cerrar valores determinados por el tiempo de construcción del árbol kd y la preparación de datos para la GPU. Además, el tiempo de construcción de un árbol contribuye significativamente al tiempo total paraλ c r i t ≤ 4 , entonces este tiempo puede ser descuidado. Es interesante notar que cuandoN = 128 K el rendimiento mejora nuevamente cuando se alcanzaλ c r i t = 1024 , lo más probable es que esto se deba al hecho de que todos los hilos de la GPU rodean los mismos vértices de árbol al mismo tiempo, lo que mejora el uso de caché L1 y elimina por completo la'divergencia de ramificación'. También se ve que el mejor rendimiento para una implementación sin una pila (cuda + heap + nested + tex + stackless), supera la versión con la pila aproximadamente1.4 - 1.5 veces. Otras implementaciones son varias veces más lentas. Para consolidar el resultado, calcularemos el tiempo en una GPU con una arquitectura más nueva.Resultados de lanzamiento en GeForce GTX 1080 Ti (precisión única)7. ( ) fn GPU N=1M λcrit=4| / | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 1024 |
|---|
| opencl+dense | 47.8 | 23.4 | 11.6 | 11.59 | 12.8 | 12.8 | 12.8 | 12.8 |
| cuda+dense | 49.0 | 23.8 | 11.9 | 11.8 | 11.7 | 11.7 | 11.7 | 11.7 |
| opencl+heap+cycle | 7.48 | 8.26 | 7.73 | 7.36 | 7.33 | 7.27 | 7.25 | 7.26 |
| opencl+heap+nested | 1.33 | 1.20 | 1.41 | 2.46 | 2.53 | 2.49 | 2.44 | 2.47 |
| cuda+heap+nested | 1.23 | 1.10 | 1.31 | 2.28 | 2.29 | 2.28 | 2.23 | 2.25 |
| cuda+heap+nested+tex | 0.88 | 0.68 | 0.654 | 1.6 | 1.6 | 1.6 | 1.6 | 1.6 |
| cuda+heap+nested+tex+stackless | 0.71 | 0.47 | 0.45 | 0.43 | 0.43 | 0.42 | 0.41 | 0.395 |

|
12. fn ( λcrit ) GPU
|
Cuando se usa la GeForce GTX 1080 Ti para el cálculo, la diferencia entre las caminatas de árbol con una pila y sin pila es hasta dos veces, siempre que descuidemos el tiempo que lleva construir el árbol. Este hecho nos empuja a garantizar que en el futuro se transfiera a la GPU y se cree un árbol. Una comparación de las tablas 5-7 muestra que no hay un único tamaño de bloque óptimo para un número diferente de cuerpos, y aún más para diferentes arquitecturas de GPU, además, la diferencia de tiempo de cálculo puede alcanzar varias veces, incluso si no tiene en cuenta los valores límite. Por lo tanto, antes de largos cálculos, tiene sentido determinar el tamaño de bloque óptimo para cada configuración.Lo principal que hemos logrado es la capacidad de modelar la interacción por pares de poco más de un millón de cuerpos (2 20 ) por un tiempo razonable en uno, no la GPU más nueva. En las GPU más nuevas (Tesla V100), aparentemente, será posible procesar alrededor de dos millones de cuerpos que interactúan en un tiempo razonable, porque su rendimiento es aproximadamente cuatro veces mayor que la mitad del Tesla K80. Aunque este número también es incomparable con el número de estrellas incluso en galaxias enanas, como laPequeña Nube de Magallanes, pero, en mi opinión, es impresionante.Conclusión
El uso de métodos integrados para resolver ecuaciones diferenciales permite resolver problemas similares con un alto nivel de precisión a un costo de tiempo relativamente pequeño, y la aplicación de algoritmos para aproximar la función de calcular las fuerzas de atracción por pares nos permite procesar volúmenes colosales de cuerpos que interactúan. Por lo tanto, a diferencia de los extraterrestres de las "Tareas de tres cuerpos", podemos resolver el problemaN cuerpos, tanto para un pequeño número de cuerpos como para grupos de estrellas enteros, aunque a costa de alguna pérdida de precisión.Visualización
Para aquellos que han leído hasta el final, les daré algunos videos más con visualización de la evolución de los sistemas de los cuerpos.Simulación de una colisión de dos galaxias. El número total de cuerpos es de 60 mil.Modelando la evolución de una galaxia que consiste en un millón de estrellas. En el centro hay un cuerpo con una masa del 99% del total. Las partículas individuales son casi indistinguibles. Ya más como una ola en una gota de líquido. Coloreado según el módulo de velocidad. Baja velocidad - azul, medio - verde, alto - rojo. Se ve que en el centro la velocidad es mayor, y disminuye gradualmente hasta los bordes, y la más baja en el plano ecuatorial.Una simulación más 'dinámica' de la evolución de una galaxia de un millón de estrellas. En el centro hay un cuerpo con una masa del 10% del total. El cuerpo central afecta el resto significativamente menos. Primero, las "estrellas" se separan, luego se reúnen en varios grupos y al final nuevamente forman un gran grupo.En el proceso de modelado, alrededor del 5% de las "estrellas" abandonaron la región inicial de manera irrevocable.En el décimo segundo, se parece mucho a la galaxia de volteretas existente .El código del proyecto se puede encontrar en el github .