Antes de comenzar la presentación del material, me gustaría decir algunas palabras sobre mí: un miembro de las comunidades contra la negación del VIH por el VIH (“disidencia del VIH / SIDA”): en 2016-2018, “Disidentes del VIH / SIDA y sus hijos”, a partir de 2018 - "Negación del VIH / SIDA y medicina alternativa".
Mi opinión, y no solo la mía, es que la mayoría de los casos de rechazo al tratamiento del VIH son causados por un malentendido banal de que se trata de una infección crónica controlada, así como por la estigmatización de las personas que viven con el VIH: el uso del cliché común de que el VIH - una enfermedad de las capas más bajas de la sociedad o viceversa, la "élite cultural". Esto no es así durante mucho tiempo: en Rusia, aproximadamente el 1% de la población vive con el VIH, y la situación no planea mejorar.
Hace aproximadamente un año, varios artículos sobre este recurso me animaron a escribir cinco notas sobre la historia de la lucha contra los virus. El propósito de estos artículos fue describir los principios de funcionamiento de varios tipos de medicamentos contra el VIH (los consultores eran un microbiólogo y un especialista en enfermedades infecciosas). Espero que disfrutes arreglando estas notas.
Acerca de los virus
Entonces, algunas palabras sobre los virus en general: ocupan una posición intermedia entre el mundo vivo y el no vivo; son incapaces de reproducción independiente; para esto, se requieren las células del organismo huésped.
El virus está organizado de manera bastante simple: lleva un código genético, el código está cerrado en una cápside, la cápsida a veces está rodeada por un caparazón. El código se puede presentar de varias maneras. El portador del código es ADN o ARN, es decir ácido nucleico (NK). Las cadenas de código pueden ser una o dos: NK bicatenario y monocatenario. La cadena se puede cerrar en un anillo o lineal. En 1971, David Baltimore, de acuerdo con estos signos, dividió los virus en 7 clases. Esta clasificación todavía está en uso hoy y será importante para explicar cómo funcionan algunos medicamentos.
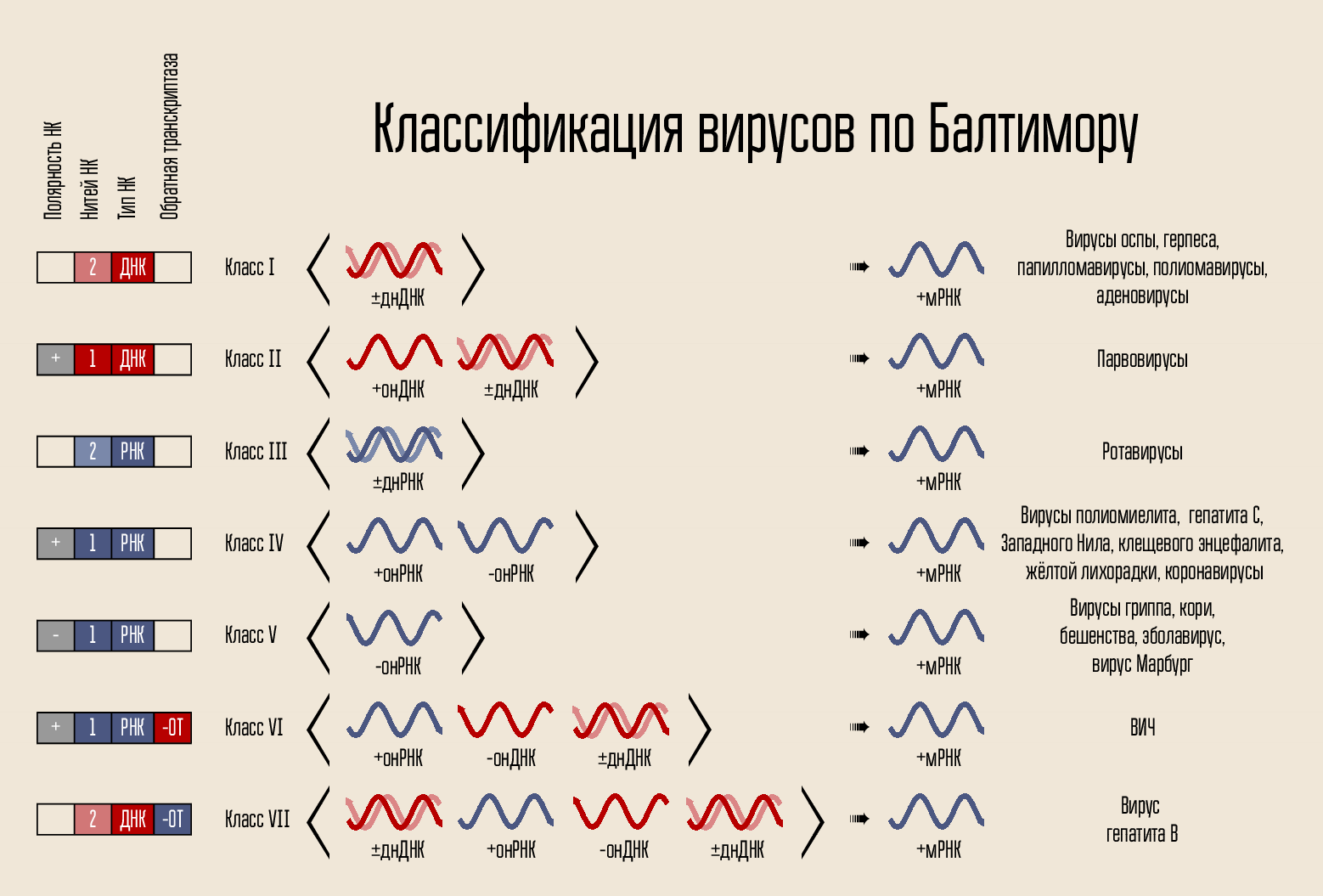
El código en sí mismo para construir un nuevo virus no puede penetrar la célula en sí misma; necesita algún mecanismo de penetración. Por lo tanto, hay una cubierta de proteína, una cápside, que protege el NK del virus y ayuda a penetrar en la célula. En algunos casos, los virus pueden tener membranas lipídicas adicionales.
La penetración del virus en la célula.

Para ingresar a una célula, el virus debe conectarse a su membrana. Para hacer esto, hay proteínas en la superficie del virus que se unen a las proteínas receptoras de la célula huésped, en lugares de la superficie de la pared celular a los que el virus puede unirse. Y deben ajustarse estrictamente al virus, de lo contrario, ni siquiera podrán adherirse a la célula.
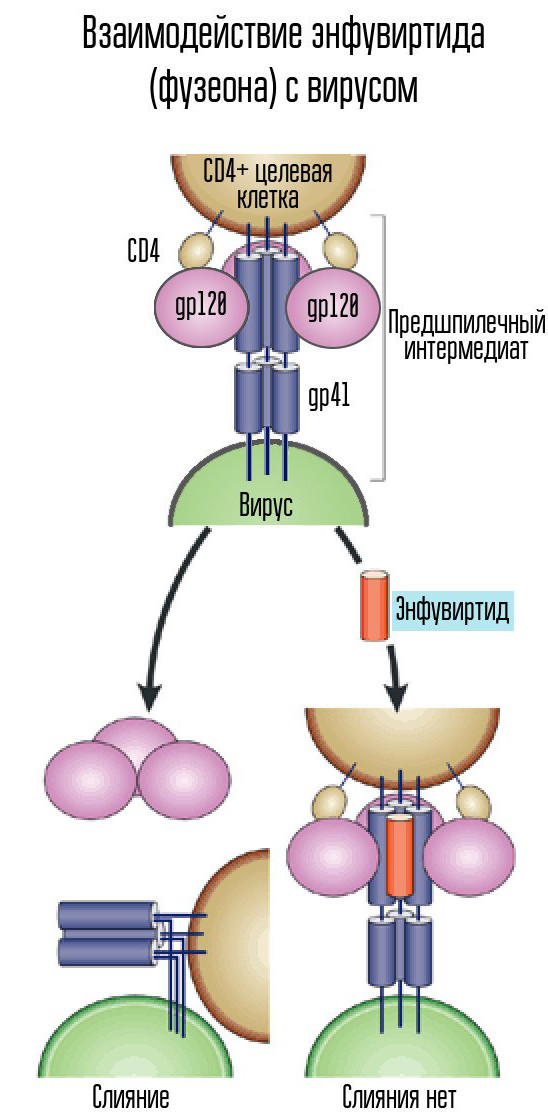
Para esto, el VIH usa el receptor CD4 (las células con dicho receptor son las células inmunes del cuerpo, incluidos los linfocitos T, monocitos, macrófagos, etc.). El CD4 solo no es suficiente: necesita otro coreceptor, CCR5 o CXCR4. El VIH usa la proteína de la envoltura gp120 para unirse. Después de esto, la forma de otra proteína de la envoltura del virus, gp41, cambia. Se dobla hacia un lado, formando una horquilla y permitiendo que la cápside del virus se fusione con la célula.
Enfuvirtida (Fuzeon), un inhibidor de la proteína gp41, es uno de los agentes utilizados para combatir el virus. Enfuvirtida se combina con esta proteína para prevenir la formación de horquillas. Por lo tanto, la cápside del virus no puede fusionarse con la célula y no se produce infección. Este medicamento es el único inhibidor de fusión desarrollado y aprobado.
Los retrovirus, a los que pertenece el VIH, son un objetivo extremadamente inconveniente para los medicamentos debido a su variabilidad. Las células humanas son mucho menos volátiles. Se sabe que aproximadamente el 1% de la población del norte de Europa es inmune al VIH: son portadores de la mutación CCR5-∂32, lo que hace que el receptor CCR5 no sea adecuado para combinar con el VIH.
Desafortunadamente, cambiar la forma de este receptor para siempre, incluso para las nuevas células que continúan apareciendo en el cuerpo humano, es una tarea extremadamente difícil (aunque ha habido intentos), pero el desarrollo de un inhibidor del receptor es un medicamento que se uniría al receptor de la célula y evitaría que el VIH se una. para él, posiblemente.
Se estaban desarrollando varios inhibidores de los receptores CCR5 y CXCR4, pero el único aprobado hasta la fecha es maraviroc, un inhibidor de CCR5.
Transcripción inversa
¿Qué sucede después de la fusión del virus con la célula en el caso del VIH?
El VIH es el virus de clase VI de Baltimore; almacena su genoma en el ARN. Hay ADN en el núcleo de la célula, por lo que el VIH necesita convertir un ácido nucleico en otro. Tal transcripción (NK → NK) se lleva a cabo mediante las enzimas correspondientes llamadas polimerasas. Para la ADN polimerasa dependiente de ARN (es decir, leer información del ARN) (es decir, a la salida de la cual aparece el ADN) hay un nombre especial: transcriptasa inversa. La transcriptasa inversa toma el desoxinucleósido deseado (por simplicidad, en realidad implica desoxinucleósido trifosfato) y construye ADN que es complementario al ARN viral correspondiente.
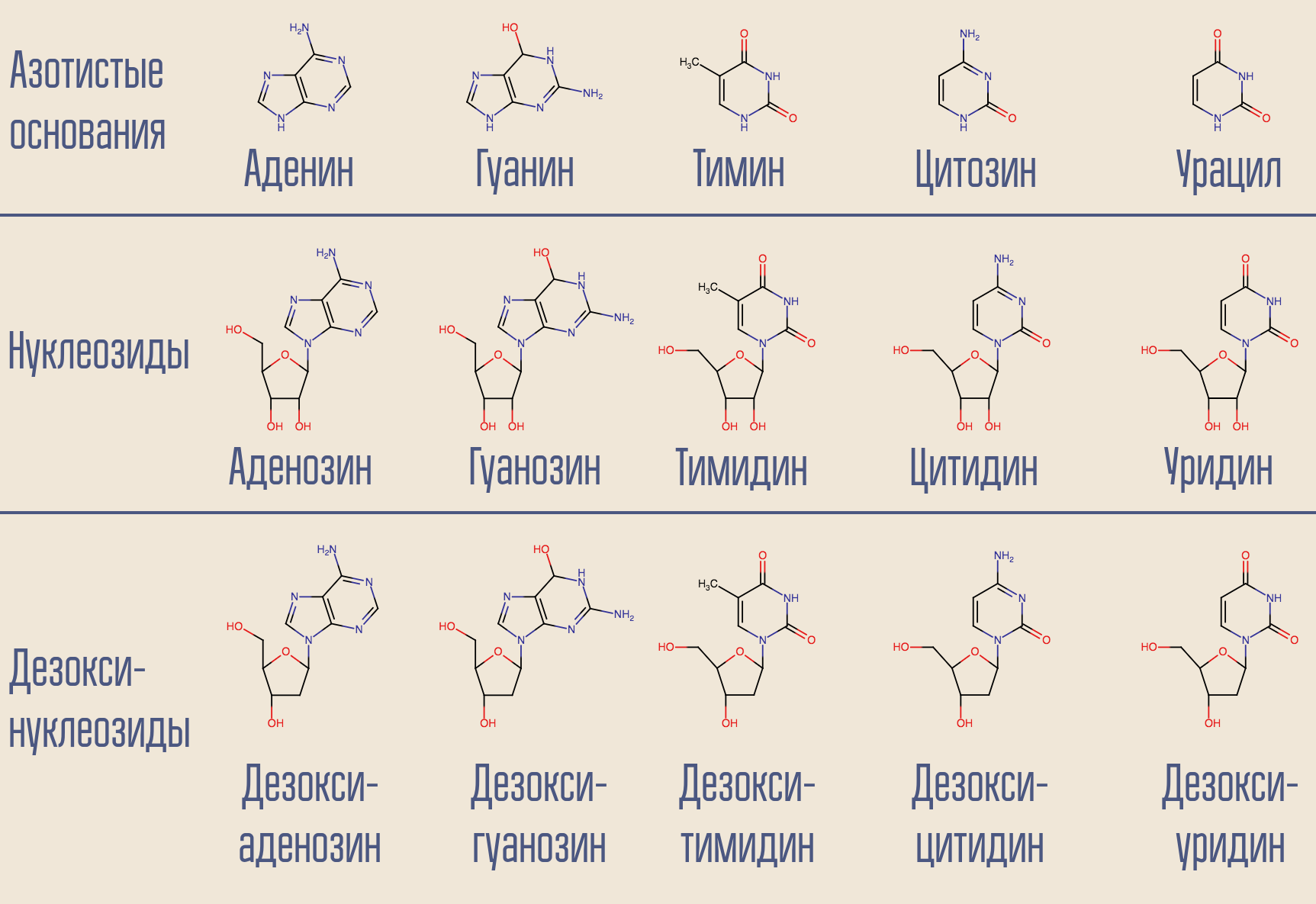
¿Es posible interrumpir este proceso? Sí, para esto solo necesita aplicar una transcriptasa inversa a algo que se parezca a un desoxinucleósido pero que no lo es. Así es como actuó el primer medicamento contra el VIH, la zidovudina (azidotimidina, AZT). Es similar a la desoxitimidina, pero no lo es.
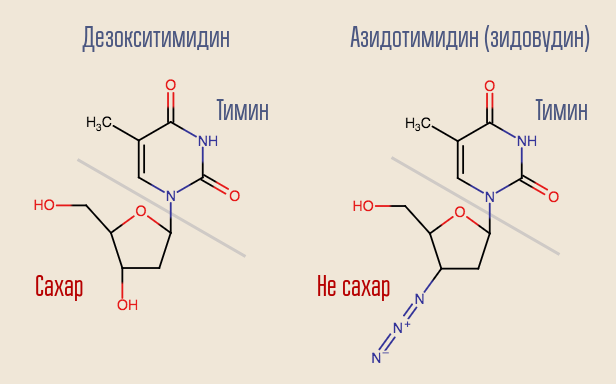
La azidotimidina se desarrolló como parte de una búsqueda de sustancias que pudieran combatir los tumores. Se suponía que estaría incrustado en la construcción del ADN humano ordinario, interrumpiéndolo. Por lo tanto, el medicamento afectaría más fuertemente a las células que se dividen más rápidamente: las células tumorales. Había ciertas razones para pensarlo: sintetizar previamente otro medicamento de este grupo, la 6-mercaptopurina, fue eficaz en el tratamiento de la leucemia.
Desafortunadamente, en las pruebas con animales, el medicamento resultó ineficaz y se olvidó por un tiempo, hasta que en 1984 el virólogo Marty St. Clair, que trabajaba en los laboratorios de la Fundación Burroughs Wellcome, inició estudios para verificar el tratamiento de todas las sustancias disponibles. tienen una nueva enfermedad: infección por VIH.
La transcriptasa inversa "reconoció" la zidovudina como desoxitimidina y trató de incorporarla al ADN. La síntesis de ADN en este sitio se interrumpió porque el medicamento solo era similar a la desoxitimidina. La zidovudina suprimió por completo la reproducción del virus, y los ensayos en humanos se iniciaron casi de inmediato.
Los voluntarios infectados por el VIH se dividieron en dos grupos, uno de los cuales recibió un placebo y el otro recibió AZT. La diferencia entre los dos grupos fue tan sorprendente que las pruebas adicionales se consideraron inhumanas: el fármaco mostró una eficacia sorprendente.
El éxito de la zidovudina ha impulsado los estudios de otros inhibidores de la transcriptasa inversa nucleósidos (INTI), y muchas otras drogas han aparecido en poco tiempo. El más interesante de los primeros medicamentos es la lamivudina, un análogo de otro desoxinucleósido, la desoxicitidina. La desventaja de la lamivudina es que con la monoterapia con este medicamento, la resistencia se desarrolla muy rápidamente, en aproximadamente un mes. Esto se debe a la mutación de un solo punto del VIH, M184V. A pesar de esto, era deseable dejar lamivudina en el esquema. El hecho es que un virus con esta mutación es hipersensible a la zidovudina, y la mutación misma reduce la tasa de replicación del virus.
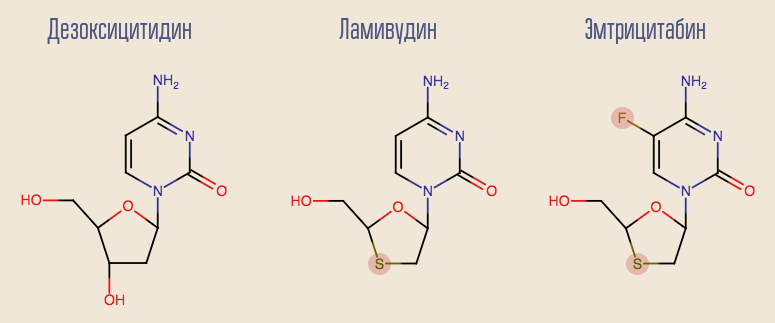
Actualmente, la lamivudina está comenzando a retroceder gradualmente hacia el pasado, dando paso a su análogo más moderno, la emtricitabina. Tanto lamivudina como el análogo de desoxiadenosina, adefovir, muestran buenos resultados en el tratamiento de la hepatitis B. Lamentablemente, se ha demostrado que adefovir no es eficaz en el tratamiento del VIH. Sin embargo, después de una ligera modificación de su molécula, apareció una versión actualizada: tenofovir. Tenofovir y emtricitabina son parte de muchas líneas modernas de terapia.
La combinación de dos NRTI podría extender significativamente la vida de las personas que viven con el VIH, sin embargo, estaba claro que para suprimir completamente el virus, era necesario incluir al menos un medicamento de un tipo diferente de acción, porque tarde o temprano el virus desarrolló resistencia a cualquier combinación de NRTI. Una de las primeras sustancias de un tipo diferente de acción fue otro tipo de inhibidor de la transcriptasa inversa: no nucleósido (NNRTI). Aunque la transcriptasa inversa (RT) quiere trabajar con algo similar a un nucleósido (nucleósido trifosfato), puede intentar hacer una sustancia que se una a RT y cambie su forma para que ya no pueda realizar sus funciones.
En 1996 y 1998, se aprobaron dos de estas sustancias, nevirapina y efavirenz, respectivamente. Cada uno de ellos suprime efectivamente el trabajo de OT, y en combinación con dos NRTI, crea un esquema completo de terapia antirretroviral altamente activa (TARGA), suficiente para que una persona que vive con VIH viva una vida plena, cuya duración no difiere mucho de la vida de una persona sin VIH .
En 2006, se aprobó el primer medicamento combinado para un solo uso por día: Atripla. Atripla consta de dos NRTI, emtricitabina y tenofovir (en forma de tenofovir disoproxilo, un profármaco, una forma de dosificación modificada químicamente que se convierte en un medicamento directamente en el cuerpo), y un NNRTI, efavirenza. Atripla se ha convertido en un paso cualitativamente nuevo para mejorar la calidad de vida de los pacientes. Hasta la fecha, los medicamentos genéricos de Atripla son uno de los medicamentos más utilizados en el mundo (en países en desarrollo).
Sin embargo, hoy los NNRTI están abandonando gradualmente el mercado: los medicamentos antiguos causan diversos efectos secundarios. Entonces, por ejemplo, los primeros dos meses después del comienzo de tomar efavirenz pueden causar mareos y otros efectos similares en algunos pacientes (¡no en todos!). Por supuesto, esto es mucho mejor que la muerte inminente; Esta condición no dura tanto tiempo, y ya han aprendido cómo lidiar con ella; sin embargo, la tendencia actual es la transición a tales medicamentos, el paciente no nota ningún efecto secundario de ellos.
Integración
Si la transcriptasa inversa ha hecho su trabajo, ¿es posible detener la incorporación de ADN viral en el ADN celular? Una enzima especial llamada integrasa está involucrada en este proceso.
El proceso de integración del ADN viral continúa en varias etapas. Inicialmente, la integrasa se une al ADN viral, eliminando el dinucleótido GT del extremo 3 'de cada cadena. Luego, todo el complejo se transporta al núcleo, donde la integrasa cataliza la etapa de transferencia de la cadena. Esta etapa es una reacción de transesterificación (intercambio de radicales): los nucleótidos de ADN de la célula se conectan no entre sí, sino con los nucleótidos del ADN viral. La integrasa ataca los enlaces internucleotídicos ubicados a una distancia de cinco nucleótidos. Así, después de la integración, queda: el procesamiento de los extremos 5 'de las cadenas de ADN viral, la finalización de 5 nucleótidos faltantes y la ligadura (la conexión de dos cadenas NK con una enzima ligasa), que se realizan con la participación de proteínas celulares [1].
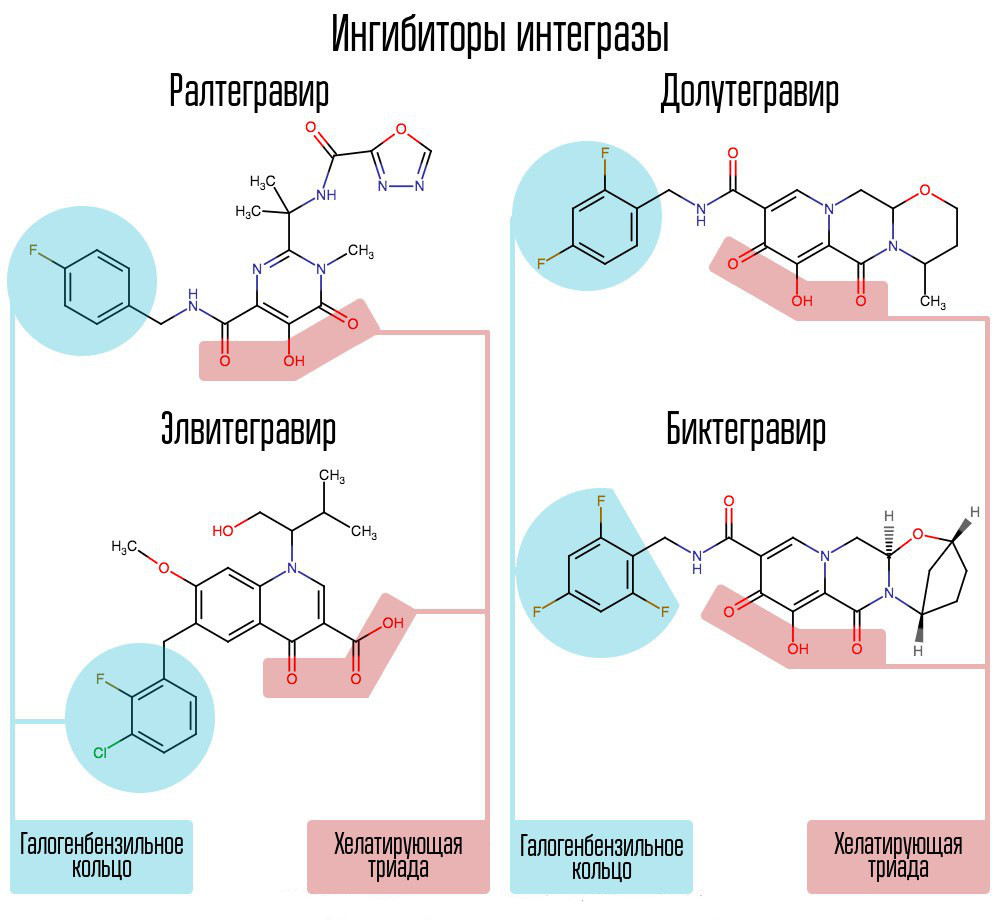
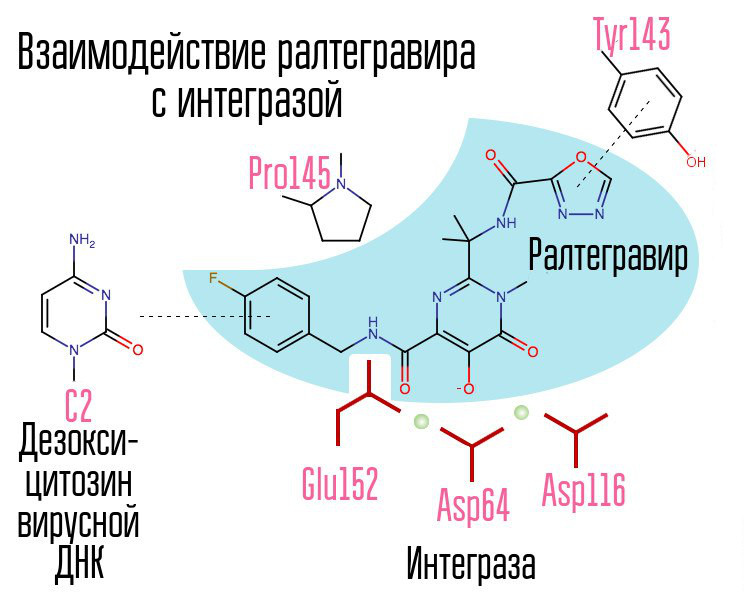
El examen de aproximadamente 250,000 sustancias en bibliotecas de compuestos químicos reveló sustancias que inhibirían la integrasa del VIH. Todos resultaron ser compuestos de ácido 2,4-dioxobutanoico. Coordinaron iones metálicos en el centro activo de integrasa, en esa parte, que era responsable de la transferencia de la cadena. Otros intentos de desarrollar inhibidores de la integrasa del VIH-1 condujeron a la aparición de un derivado de la N-pirimidinona, una sustancia MK-0518, llamada raltegravir. [2]
Común a raltegravir e inhibidores posteriores de la integrasa son la tríada quelante (iones metálicos de coordinación) y el anillo de halógeno-bencilo que interactúa con la penúltima desoxicitosina en el extremo 3 'del ADN viral unido a la enzima.
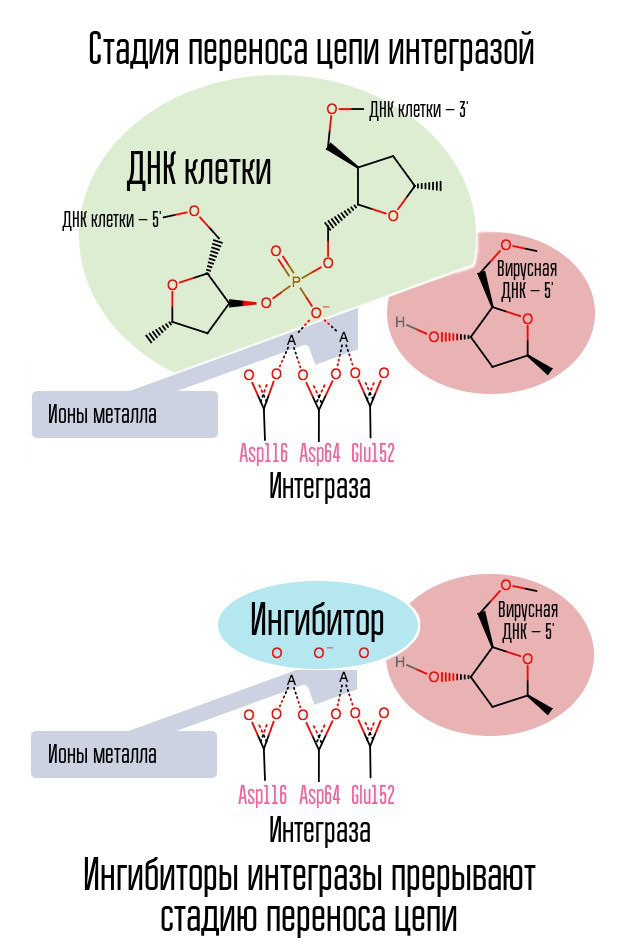
El proceso de integración del virus en la célula es el último paso en el que la profilaxis posterior a la exposición es efectiva. Después de eso, las células que transportan el ADN del VIH en su núcleo aparecen en el cuerpo humano. La ventana más efectiva para la profilaxis posterior a la exposición es de aproximadamente 6-10 horas.
El anillo de halógenobencilo en la molécula inhibidora de la integrasa interactúa con el ADN viral, y el grupo de átomos de oxígeno interactúa con dos átomos metálicos. La integrasa del virus utiliza estos átomos metálicos para introducir ADN viral en la célula. Como resultado, el proceso de integración está bloqueado.
Las IA modernas, como dolutegravir, han podido derrotar las "enfermedades infantiles" de raltegravir asociadas con la rápida formación de resistencia.
Proteólisis
Después de que el genoma viral pasa la etapa de transcripción, los ARN virales creados se envían para salir de la célula. En el proceso de creación del virión, está involucrada otra enzima viral llamada proteasa. La proteasa corta poliproteínas largas en proteínas funcionales individuales, lo que resulta en la formación de enzimas virales y proteínas estructurales del virus.
La proteasa es activa no solo contra las proteínas del VIH, sino también contra las proteínas de la célula huésped, lo que puede explicar el efecto citotóxico del VIH (muerte celular).
Si la proteasa está bloqueada, el virión no podrá pasar por la etapa de maduración y permanecerá completamente no funcional. La proteasa del VIH-1 es una proteasa aspártica retroviral típica que tiene la secuencia de aminoácidos característica Asp25 Thr26 Gly27 (ácido aspártico - treonina - glicina) en el centro activo. El primer inhibidor de la proteasa, saquinavir, fue aprobado por la FDA el 6 de diciembre de 1995. Por lo tanto, fue después de la creación de saquinavir que la terapia antirretroviral altamente activa estuvo disponible por primera vez.
Otro representante típico de este grupo de medicamentos es el lopinavir (utilizado junto con ritonavir, kaletra, uno de los medicamentos contra el VIH más comunes en Rusia). El ritonavir también es un inhibidor de la proteasa, pero se usa como refuerzo; gracias a su acción, aumenta la concentración del fármaco principal.
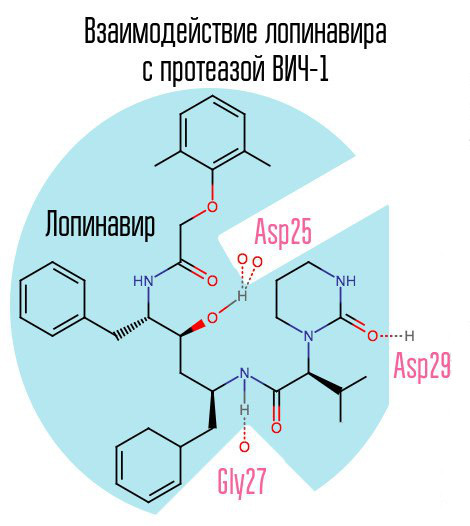
Dado que el saquinavir y los inhibidores de proteasa (IP) posteriores están dirigidos específicamente al centro activo de la enzima, con el desarrollo de resistencia a un IP, existe una alta probabilidad de que ocurra resistencia a otros IP. La solución a este problema puede ser la creación de tales inhibidores que se dirigen a otras zonas de proteasa.
El medicamento darunavir (prezista), que apareció en 2006, alivió de alguna manera el problema de las cepas de VIH-1 resistentes a la IP, ya que formó un vínculo no utilizado previamente con ácido aspártico en la posición 30.
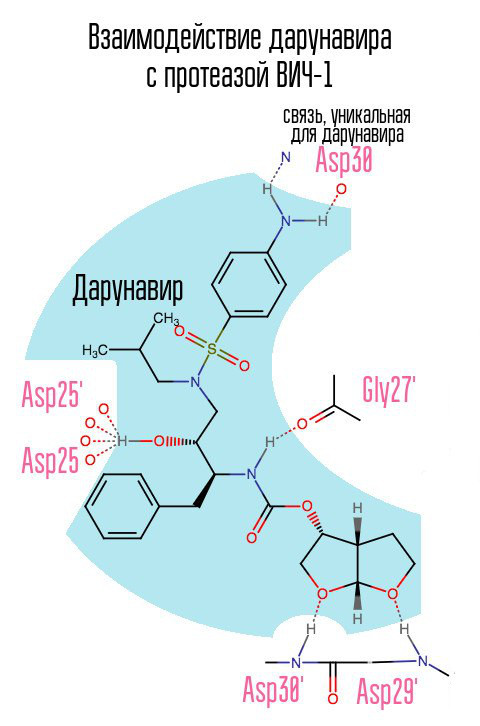
Sin proteasa, el virus no puede someterse al proceso de maduración. Los inhibidores se unen al centro activo de la proteasa y evitan que funcione.
Los inhibidores de la proteasa son altamente efectivos con alta carga viral: dado que en este momento nacen muchos nuevos viriones en el cuerpo, los IP no les permiten madurar, lo que reduce efectivamente la carga viral en poco tiempo. Sin embargo, por el momento, los IP no se utilizan en la terapia de primera línea, dando paso a los inhibidores de la integrasa (II).
La razón de esto fueron los efectos secundarios: el hecho es que, por ejemplo, la misma kaletra condujo a una inhibición inespecífica de la proteólisis de proteínas suministradas con alimentos, como resultado de lo cual estas proteínas ingresaron al intestino delgado y causaron diarrea. Seguir una dieta específica o usar nuevos IP, como el prezista, puede reducir este efecto a casi cero, pero otro efecto asociado con el aumento de los niveles de azúcar a menudo impide el uso indefinido de inhibidores de la proteasa.
Regímenes de tratamiento modernos
Hasta la fecha, los más modernos se consideran esquemas que consisten en un inhibidor de la integrasa y uno o dos INTI (dolutegravir + abacavir + lamivudina; dolutegravir + lamivudina es un esquema popular de dos componentes, sin embargo, no es adecuado para todos). Estos esquemas permiten a una persona vivir una vida plena que no difiere en duración de la vida de una persona sin VIH.
A pesar de todos los éxitos, todavía no es posible una cura completa para el VIH (el trasplante de células madre de médula ósea de un donante con mutación CCR5-∂32 permite lograr ese resultado, pero, aparentemente, solo si ocurre una reacción de trasplante versus huésped, en un gran número de casos que conducen a la muerte del destinatario).
Conclusión
Los métodos desarrollados para el VIH han ayudado en la lucha contra otras enfermedades infecciosas: como se mencionó anteriormente, la lamivudina y el tenofovir son efectivos contra el virus de la hepatitis B (Baltimore clase VII - la polimerasa de la hepatitis B puede transferir ARN al ADN, por lo que algunos NRTI son eficaces para combatirlo) . El conocimiento adquirido ayudó a desarrollar medicamentos de acción directa contra la hepatitis C, que hoy pueden curar por completo esta enfermedad (la hepatitis C no tiene una fase latente, por lo tanto, al suprimir la carga viral, los nuevos viriones no tienen de dónde venir: la enfermedad está completamente curada).
[1] Korolev S. P., Agapkina Yu. Yu., Gottikh M. B. Problemas y perspectivas del uso clínico de los inhibidores de la integración del VIH-1
[2] Shahgildyan V.I. Inhibidores de la integrasa del VIH: la base de una terapia antirretroviral efectiva y segura