¿Qué sucede si tiene una idea para una proteína fresca y saludable y quiere obtenerla en realidad? Por ejemplo, ¿le gustaría crear una vacuna contra
H. pylori (como el
equipo esloveno en iGEM 2008 ) creando una proteína híbrida que combine fragmentos de flagelina de
E. coli que estimulen la respuesta inmune con la flagelina de
H. pylori habitual?
H. Pylori Hybrid Flagellin Design presentado por el equipo esloveno en iGEM 2008Sorprendentemente, estamos muy cerca de crear cualquier proteína que queramos sin abandonar el portátil Jupyter, gracias a los últimos desarrollos en genómica, biología sintética y, más recientemente, en laboratorios en la nube.
En este artículo, mostraré el código de Python desde la idea de una proteína hasta su expresión en una célula bacteriana, sin tocar una pipeta ni hablar con nadie. ¡El costo total será de unos pocos cientos de dólares! Usando la
terminología de Vijaya Pande de A16Z , esto es Biología 2.0.
Más específicamente, en el artículo, el código Python del laboratorio de la nube hace lo siguiente:
- Síntesis de una secuencia de ADN que codifica cualquier proteína que quiera.
- Clonando este ADN sintético en un vector que puede expresarlo.
- Transformación de bacterias con este vector y confirmación de que está ocurriendo la expresión.
Configuración de Python
Primero, la configuración general de Python que se necesita para cualquier bloc de notas Jupyter. Importamos algunos módulos Python útiles y creamos algunas funciones de utilidad, principalmente para la visualización de datos.
Códigoimport re import json import logging import requests import itertools import numpy as np import seaborn as sns import pandas as pd import matplotlib as mpl import matplotlib.pyplot as plt from io import StringIO from pprint import pprint from Bio.Seq import Seq from Bio.Alphabet import generic_dna from IPython.display import display, Image, HTML, SVG def uprint(astr): print(astr + "\n" + "-"*len(astr)) def show_html(astr): return display(HTML('{}'.format(astr))) def show_svg(astr, w=1000, h=1000): SVG_HEAD = '''<?xml version="1.0" standalone="no"?><!DOCTYPE svg PUBLIC "-//W3C//DTD SVG 1.1//EN" "http://www.w3.org/Graphics/SVG/1.1/DTD/svg11.dtd">''' SVG_START = '''<svg viewBox="0 0 {w:} {h:}" version="1.1" xmlns="http://www.w3.org/2000/svg" xmlns:xlink= "http://www.w3.org/1999/xlink">''' return display(SVG(SVG_HEAD + SVG_START.format(w=w, h=h) + astr + '</svg>')) def table_print(rows, header=True): html = ["<table>"] html_row = "</td><td>".join(k for k in rows[0]) html.append("<tr style='font-weight:{}'><td>{}</td></tr>".format('bold' if header is True else 'normal', html_row)) for row in rows[1:]: html_row = "</td><td>".join(row) html.append("<tr style='font-family:monospace;'><td>{:}</td></tr>".format(html_row)) html.append("</table>") show_html(''.join(html)) def clean_seq(dna): dna = re.sub("\s","",dna) assert all(nt in "ACGTN" for nt in dna) return Seq(dna, generic_dna) def clean_aas(aas): aas = re.sub("\s","",aas) assert all(aa in "ACDEFGHIKLMNPQRSTVWY*" for aa in aas) return aas def Images(images, header=None, width="100%"):
Laboratorios de nubes
Al igual que AWS o cualquier nube informática, el laboratorio de la nube cuenta con equipos de biología molecular, así como con robots que alquila a través de Internet. Puede emitir instrucciones a sus robots haciendo clic en algunos botones en la interfaz o escribiendo código que los programe usted mismo. No es necesario escribir sus propios protocolos, como haré aquí, una parte importante de la biología molecular son las tareas de rutina estándar, por lo que generalmente es mejor confiar en un protocolo alienígena confiable que muestre una buena interacción con los robots.
Recientemente, han aparecido varias compañías con laboratorios en la nube:
Transcriptic ,
Autodesk Wet Lab Accelerator (beta, y construido sobre la base de Transcriptic),
Arcturus BioCloud (beta),
Emerald Cloud Lab (beta),
Synthego (aún no ha comenzado). Incluso hay empresas creadas sobre laboratorios en la nube, como
Desktop Genetics , que se especializa en CRISPR. Están comenzando a aparecer
artículos científicos sobre el uso de los laboratorios en la nube en la ciencia real.
Al momento de escribir este artículo, solo Transcriptic está en el dominio público, por lo que lo usaremos. Según tengo entendido, la mayor parte del negocio de Transcriptic se basa en la automatización de protocolos comunes, y escribir sus propios protocolos en Python (como haré en este artículo) es menos común.
 Transcriptic "celda de trabajo" con refrigeradores en la parte inferior y diversos equipos de laboratorio en el stand
Transcriptic "celda de trabajo" con refrigeradores en la parte inferior y diversos equipos de laboratorio en el standDaré instrucciones a los robots transcriptos sobre el
protocolo automático . Autoprotocol es un lenguaje basado en JSON para escribir protocolos para robots de laboratorio (y humanos, por así decirlo). Autoprotocol se realiza principalmente en
esta biblioteca de Python . El lenguaje fue creado originalmente y todavía es compatible con Transcriptic, pero, según tengo entendido, está completamente abierto. Hay buena
documentación .
Una idea interesante es que en el protocolo automático puede escribir instrucciones para personas en laboratorios remotos, por ejemplo, en China o India, y potencialmente obtener algunas ventajas al usar tanto personas (su juicio) como robots (falta de juicio). Necesitamos mencionar los
protocolos. Aquí , este es un intento de estandarizar protocolos para mejorar la reproducibilidad, pero para los humanos, no para los robots.
"instructions": [ { "to": [ { "well": "water/0", "volume": "500.0:microliter" } ], "op": "provision", "resource_id": "rs17gmh5wafm5p" }, ... ]
Ejemplo de fragmento de autoprotocoloConfiguración de Python para biología molecular
Además de importar bibliotecas estándar, necesitaré algunas utilidades biológicas moleculares específicas. Este código es principalmente para auto-protocolo y transcriptic.
El concepto de "volumen muerto" a menudo se encuentra en el código. Esto significa la última gota de líquido que los robots Transcriptic no pueden tomar con una pipeta de los tubos (¡porque no pueden verla!). Tienes que pasar mucho tiempo para asegurarte de que los matraces tengan suficiente material.
Código import autoprotocol from autoprotocol import Unit from autoprotocol.container import Container from autoprotocol.protocol import Protocol from autoprotocol.protocol import Ref
Síntesis de ADN y biología sintética
A pesar de su conexión con la biología sintética moderna, la síntesis de ADN es una tecnología bastante antigua. Durante décadas, hemos podido fabricar oligonucleótidos (es decir, secuencias de ADN de hasta 200 bases). Sin embargo, siempre fue costoso, y la química nunca permitió secuencias largas de ADN. Recientemente, ha sido posible sintetizar genes completos a un precio razonable (hasta miles de bases). Este logro realmente abre la era de la "biología sintética".
Synthetic Genomics de Craig Venter
ha llevado la biología sintética más lejos al
sintetizar un organismo completo , con más de un millón de bases de longitud. A medida que aumenta la longitud del ADN, el problema ya no es la síntesis, sino el ensamblaje (es decir, unir secuencias de ADN sintetizadas). Con cada ensamblaje, puede duplicar la longitud del ADN (o más), por lo que después de una docena de iteraciones, ¡obtendrá una
molécula bastante larga ! La distinción entre síntesis y ensamblaje pronto debería quedar clara para el usuario final.
Ley de Moore?
El precio de la síntesis de ADN está cayendo bastante rápido, de más de $ 0.30 hace un año dos a aproximadamente $ 0.10 hoy, pero se está desarrollando más como bacterias que como procesadores. En contraste, los precios de secuenciación de ADN están cayendo más rápido que la ley de Moore. Un objetivo de
$ 0.02 por base se establece como un punto de inflexión donde puede reemplazar muchas manipulaciones de ADN que consumen mucho tiempo con síntesis simple. Por ejemplo, a este precio, puede sintetizar un plásmido completo de 3 kb por
$ 60 y omitir un montón de biología molecular. Espero que lo logremos en un par de años.
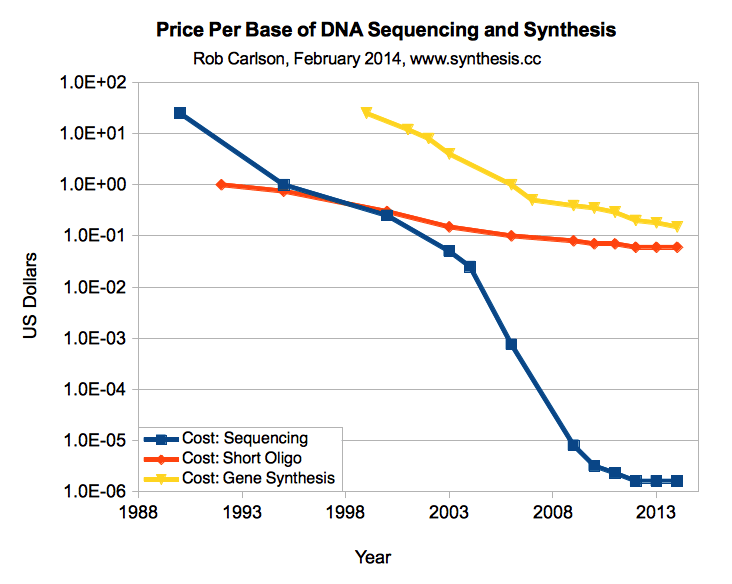 Precios de síntesis de ADN en comparación con los precios de secuenciación de ADN, precio por 1 base (Carlson, 2014)
Precios de síntesis de ADN en comparación con los precios de secuenciación de ADN, precio por 1 base (Carlson, 2014)Empresas de síntesis de ADN
Hay varias compañías grandes en el campo de la síntesis de ADN: IDT es el mayor productor de oligonucleótidos y también puede producir "fragmentos de genes" (
gBlocks ) más largos (hasta
2kb ).
Gen9 ,
Twist y
DNA 2.0 generalmente se especializan en secuencias de ADN más largas: estas son compañías de síntesis de genes. También hay algunas compañías nuevas interesantes, como
Cambrian Genomics y
Genesis DNA , que están trabajando en los métodos de síntesis de próxima generación.
Otras compañías, como
Amyris ,
Zymergen y
Ginkgo Bioworks , usan el ADN sintetizado por estas compañías para trabajar a nivel corporal.
Synthetic Genomics también hace esto, pero sintetiza el ADN mismo.
Ginkgo recientemente
llegó a un acuerdo con Twist para hacer 100 millones de bases: el acuerdo más grande que he visto. Esto demuestra que vivimos en el futuro, Twist incluso anunció un código promocional en Twitter: cuando compras 10 millones de bases de ADN (¡casi todo el genoma de la levadura!), Obtienes otros 10 millones gratis.
 Oferta de nicho de Twitter Twist
Oferta de nicho de Twitter TwistPrimera parte: diseño de experimentos
Proteína fluorescente verde
En este experimento, sintetizamos una secuencia de ADN para una
proteína fluorescente verde simple (GFP). La proteína GFP se encontró por primera vez en una
medusa que fluoresce bajo la luz ultravioleta. Esta es una proteína extremadamente útil porque es fácil detectar su expresión simplemente midiendo la fluorescencia. Hay opciones de GFP que producen amarillo, rojo, naranja y otros colores.
Es interesante ver cómo varias mutaciones afectan el color de una proteína, y este es un problema de aprendizaje automático potencialmente interesante. Más recientemente, tendrías que pasar mucho tiempo en el laboratorio para esto, ¡pero ahora te mostraré que es (casi) tan fácil como editar un archivo de texto!
Técnicamente, mi GFP es una opción de Super Carpeta (sfGFP) con algunas mutaciones para mejorar la calidad.
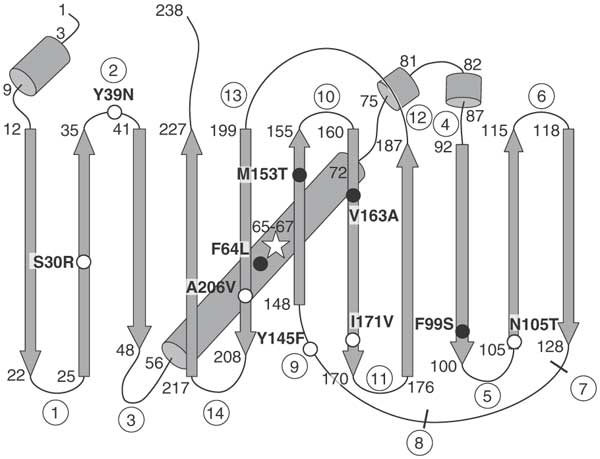 En superfolder-GFP (sfGFP), algunas mutaciones le dan ciertas propiedades útiles.
En superfolder-GFP (sfGFP), algunas mutaciones le dan ciertas propiedades útiles.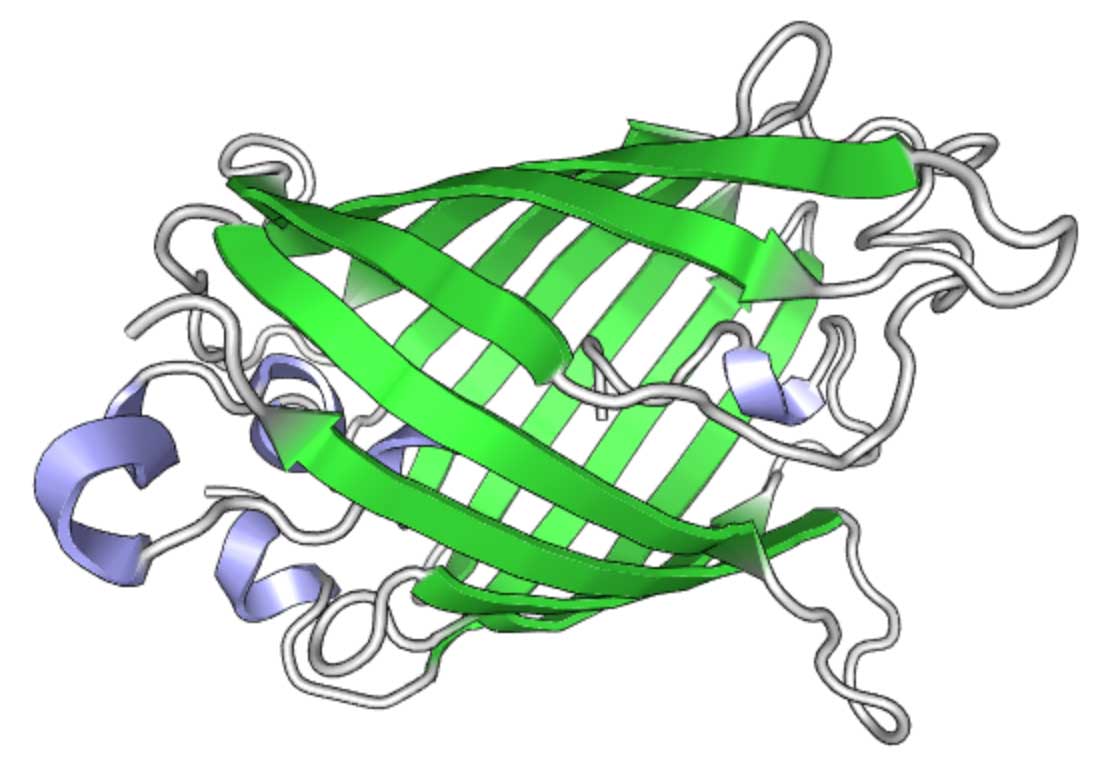 Estructura de GFP (visualizada usando PV )
Estructura de GFP (visualizada usando PV )Síntesis de GFP en Twist
Tuve la suerte de ingresar al programa de pruebas alfa de Twist, así que utilicé su servicio de síntesis de ADN (hicieron un pequeño pedido, ¡gracias Twist!). Esta es una nueva empresa en nuestro campo, con un nuevo proceso de síntesis simplificado. Sus precios rondan los
$ 0,10 por base o menos , pero
todavía están
en beta y el programa alfa en el que participé se cerró. Twist recaudó alrededor de $ 150 millones, por lo que su tecnología es animada.
Envié mi secuencia de ADN a Twist como una hoja de cálculo de Excel (todavía no hay API, pero supongo que será pronto), y enviaron el ADN sintetizado directamente a mi caja en el laboratorio Transcriptic (también usé IDT para síntesis, pero no enviaron ADN justo en Transcriptic, que estropea un poco la diversión).
Obviamente, este proceso aún no se ha convertido en un caso de uso típico y requiere algo de soporte, pero funcionó, por lo que toda la tubería permanece virtual. Sin esto, probablemente necesitaría acceso al laboratorio: muchas compañías no enviarán ADN o reactivos a su domicilio.
 GFP es inofensivo, por lo que se resalta cualquier tipo
GFP es inofensivo, por lo que se resalta cualquier tipoVector plásmido
Para expresar esta proteína en bacterias, el gen necesita vivir en algún lugar, de lo contrario, el ADN sintético que codifica el gen simplemente se degrada instantáneamente. Como regla general, en biología molecular usamos un plásmido, una pieza de ADN redondo que vive fuera del genoma bacteriano y expresa proteínas. Los plásmidos son una forma conveniente para que las bacterias compartan módulos funcionales útiles e independientes, como la resistencia a los antibióticos. Puede haber cientos de plásmidos en una célula.
La terminología ampliamente utilizada es que un plásmido es un
vector , y el ADN sintético es una inserción (inserción). Entonces, aquí estamos tratando de clonar la inserción en un vector y luego transformar la bacteria usando el vector.
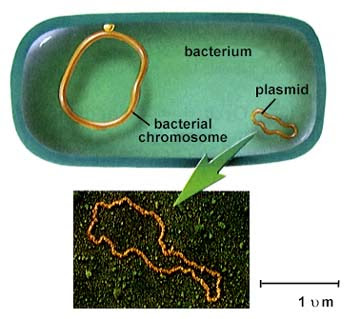 Genoma bacteriano y plásmido (¡no a escala!) ( Wikipedia )
Genoma bacteriano y plásmido (¡no a escala!) ( Wikipedia )pUC19
Elegí un plásmido bastante estándar en la serie
pUC19 . Este plásmido se usa con mucha frecuencia y, dado que está disponible como parte del inventario transcriptico estándar, no necesitamos enviarles nada.
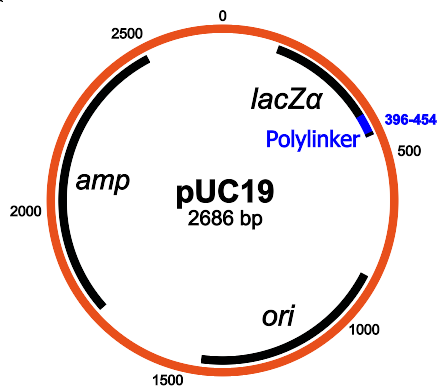 Estructura de pUC19: los componentes principales son el gen de resistencia a la ampicilina, lacZα, MCS / polylinker y el origen de la replicación (Wikipedia)
Estructura de pUC19: los componentes principales son el gen de resistencia a la ampicilina, lacZα, MCS / polylinker y el origen de la replicación (Wikipedia)PUC19 tiene una buena función: dado que contiene el gen lacZα, puede usar el método de
selección azul-blanco y ver en qué colonias la inserción fue exitosa. Se necesitan dos productos químicos:
IPTG y
X-gal , y el circuito funciona de la siguiente manera:
- IPTG induce la expresión de lacZα.
- Si lacZα se desactiva mediante el ADN insertado en el sitio de clonación múltiple ( MCS / polylinker ) en lacZα, entonces el plásmido no puede hidrolizar X-gal y estas colonias serán blancas en lugar de azules.
- Por lo tanto, una inserción exitosa produce colonias blancas y una inserción fallida produce colonias azules.
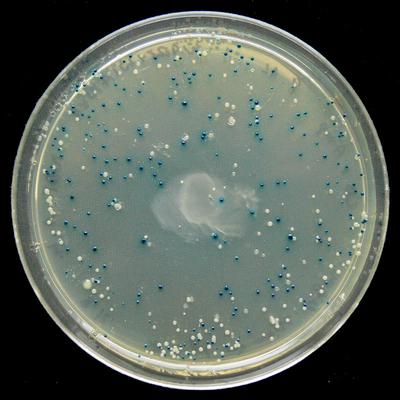 La selección azul y blanca muestra dónde se desactivó la expresión de lacZα ( Wikipedia )La documentación de openwetware
La selección azul y blanca muestra dónde se desactivó la expresión de lacZα ( Wikipedia )La documentación de openwetware dice:
E. coli DH5α no requiere IPTG para inducir la expresión del promotor lac, incluso si se expresa un represor Lac en la cepa. El número de copias de la mayoría de los plásmidos excede el número de represores en las células. Si necesita una expresión máxima, agregue IPTG a una concentración final de 1 mM.
Secuencias de ADN sintético
Secuencia de ADN de SfGFP
Es fácil obtener la secuencia de ADN para sfGFP tomando
la secuencia de proteínas y codificándola
con codones adecuados para el organismo huésped (aquí,
E. coli ). Esta es una proteína de tamaño mediano con 236 aminoácidos, por lo que a 10 centavos la síntesis de ADN cuesta alrededor de
$ 70 por base.
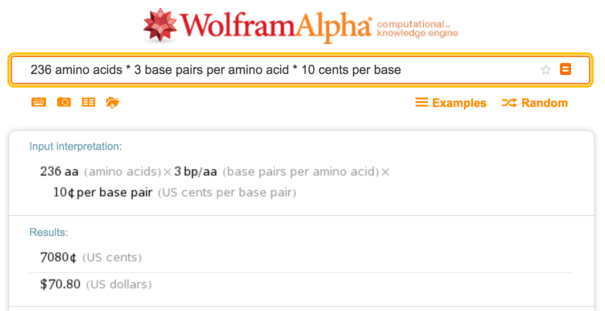 Wolfram Alpha, cálculo del costo de síntesis
Wolfram Alpha, cálculo del costo de síntesisLas primeras 12 bases de nuestro sfGFP son la
secuencia Shine-Delgarno , que agregué yo mismo, que en teoría debería aumentar la expresión (AGGAGGACAGCT, luego ATG (
codón de inicio ) inicia la proteína). De acuerdo con una herramienta computacional desarrollada por
Salis Lab (
diapositivas de la conferencia ), podemos esperar una expresión media a alta de nuestra proteína (tasa de inicio de traducción de 10,000 "unidades arbitrarias").
sfGFP_plus_SD = clean_seq(""" AGGAGGACAGCTATGTCGAAAGGAGAAGAACTGTTTACCGGTGTGGTTCCGATTCTGGTAGAACTGGA TGGGGACGTGAACGGCCATAAATTTAGCGTCCGTGGTGAGGGTGAAGGGGATGCCACAAATGGCAAAC TTACCCTTAAATTCATTTGCACTACCGGCAAGCTGCCGGTCCCTTGGCCGACCTTGGTCACCACACTG ACGTACGGGGTTCAGTGTTTTTCGCGTTATCCAGATCACATGAAACGCCATGACTTCTTCAAAAGCGC CATGCCCGAGGGCTATGTGCAGGAACGTACGATTAGCTTTAAAGATGACGGGACCTACAAAACCCGGG CAGAAGTGAAATTCGAGGGTGATACCCTGGTTAATCGCATTGAACTGAAGGGTATTGATTTCAAGGAA GATGGTAACATTCTCGGTCACAAATTAGAATACAACTTTAACAGTCATAACGTTTATATCACCGCCGA CAAACAGAAAAACGGTATCAAGGCGAATTTCAAAATCCGGCACAACGTGGAGGACGGGAGTGTACAAC TGGCCGACCATTACCAGCAGAACACACCGATCGGCGACGGCCCGGTGCTGCTCCCGGATAATCACTAT TTAAGCACCCAGTCAGTGCTGAGCAAAGATCCGAACGAAAAACGTGACCATATGGTGCTGCTGGAGTT CGTGACCGCCGCGGGCATTACCCATGGAATGGATGAACTGTATAAA""") print("Read in sfGFP plus Shine-Dalgarno: {} bases long".format(len(sfGFP_plus_SD))) sfGFP_aas = clean_aas("""MSKGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLTYG VQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNFNSHNVYITADKQKN GIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHGMDELYK""") assert sfGFP_plus_SD[12:].translate() == sfGFP_aas print("Translation matches protein with accession 532528641")
Lea en sfGFP plus Shine-Dalgarno: 726 bases de largo
La traducción coincide con la proteína con la adhesión 532528641
Secuencia de ADN PUC19
Primero, verifico que la
secuencia pUC19 que descargué del NEB tiene la longitud correcta e incluye el
polienlazador esperado.
pUC19_fasta = !cat puc19fsa.txt pUC19_fwd = clean_seq(''.join(pUC19_fasta[1:])) pUC19_rev = pUC19_fwd.reverse_complement() assert all(nt in "ACGT" for nt in pUC19_fwd) assert len(pUC19_fwd) == 2686 pUC19_MCS = clean_seq("GAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTT") print("Read in pUC19: {} bases long".format(len(pUC19_fwd))) assert pUC19_MCS in pUC19_fwd print("Found MCS/polylinker")
Leer en pUC19: 2686 bases de largo
MCS / polienlazador encontrado
Hacemos algunos QC básicos para asegurarnos de que EcoRI y BamHI estén presentes en pUC19 solo una vez (las siguientes enzimas de restricción están disponibles en el inventario transcriptico predeterminado:
PstI ,
PvuII ,
EcoRI ,
BamHI ,
BbsI ,
BsmBI ).
REs = {"EcoRI":"GAATTC", "BamHI":"GGATTC"} for rename, res in REs.items(): assert (pUC19_fwd.find(res) == pUC19_fwd.rfind(res) and pUC19_rev.find(res) == pUC19_rev.rfind(res)) assert (pUC19_fwd.find(res) == -1 or pUC19_rev.find(res) == -1 or pUC19_fwd.find(res) == len(pUC19_fwd) - pUC19_rev.find(res) - len(res)) print("Asserted restriction enzyme sites present only once: {}".format(REs.keys()))
Ahora miramos la secuencia lacZα y verificamos que no haya nada inesperado. Por ejemplo, debe comenzar con Met y terminar con un codón de parada. También es fácil confirmar que este es el ORF lacZα completo de 324 pb cargando la secuencia de pUC19 en el
visor de genes libres.
lacZ = pUC19_rev[2217:2541] print("lacZα sequence:\t{}".format(lacZ)) print("r_MCS sequence:\t{}".format(pUC19_MCS.reverse_complement())) lacZ_p = lacZ.translate() assert lacZ_p[0] == "M" and not "*" in lacZ_p[:-1] and lacZ_p[-1] == "*" assert pUC19_MCS.reverse_complement() in lacZ assert pUC19_MCS.reverse_complement() == pUC19_rev[2234:2291] print("Found MCS once in lacZ sequence")
secuencia lacZ: ATGACCATGATTACGCCAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGTAATAGCGAAGAGGCCCGCACCGATCGCCCTTCCCAACAGTTGCGCAGCCTGAATGGCGAATGGCGCCTGATGCGGTATTTTCTCCTTACGCATCTGTGCGGTATTTCACACCGCATATGGTGCACTCTCAGTACAATCTGCTCTGATGCCGCATAG
Secuencia r_MCS: AAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTC
MCS encontrado una vez en secuencia lacZ
Asamblea Gibson
Ensamblar ADN simplemente significa entrecruzar fragmentos. Por lo general, recolecta varios fragmentos de ADN en un segmento más largo y luego lo clona en un plásmido o genoma. En este experimento, quiero clonar un segmento de ADN en el plásmido pUC19 debajo del promotor lac para la expresión en
E. coli .
Existen muchos métodos de clonación (p.
Ej. ,
NEB ,
openwetware ,
addgene ). Aquí usaré el ensamblaje Gibson (
desarrollado por Daniel Gibson en Synthetic Genomics en 2009), que no es necesariamente el método más barato, sino simple y flexible. ¡Solo necesita colocar el ADN que desea recolectar (con la superposición apropiada) en un tubo de ensayo con Gibson Assembly Master Mix, y se ensamblará solo!
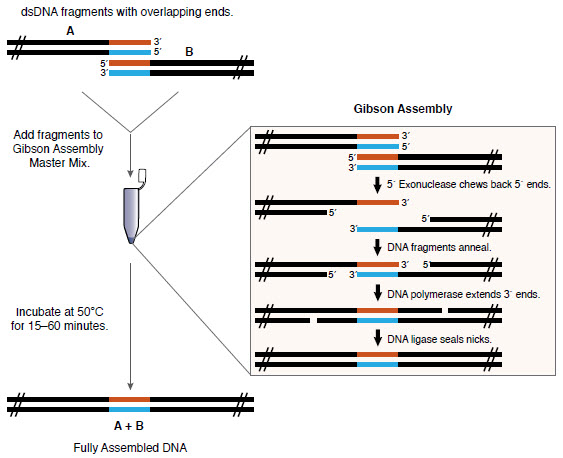 Revisión de la Asamblea de Gibson ( NEB )
Revisión de la Asamblea de Gibson ( NEB )Material de origen
Comenzamos con 100 ng de ADN sintético en 10 μl de líquido. Esto equivale a 0.21 picomoles de ADN o una concentración de 10 ng / μl.
pmol_sfgfp = convert_ug_to_pmol(0.1, len(sfGFP_plus_SD)) print("Insert: 100ng of DNA of length {:4d} equals {:.2f} pmol".format(len(sfGFP_plus_SD), pmol_sfgfp))
Inserto: 100 ng de ADN de longitud 726 equivalen a 0.21 pmol
Según
el protocolo de ensamblaje de NEB , este es suficiente material de origen:
NEB recomienda un total de 0.02-0.5 picomoles de fragmentos de ADN cuando 1 o 2 fragmentos se ensamblan en el vector, o 0.2-1.0 picomoles de fragmentos de ADN cuando se recogen 4-6 fragmentos.
0.02-0.5 pmoles * X μl
* La eficiencia de clonación optimizada es de 50-100 ng de vectores con un exceso de inserciones de 2-3 veces. Utilice 5 veces más inserciones si el tamaño es inferior a 200 bps. El volumen total de fragmentos de PCR sin filtrar en la reacción de ensamblaje de Gibson no debe exceder el 20%.
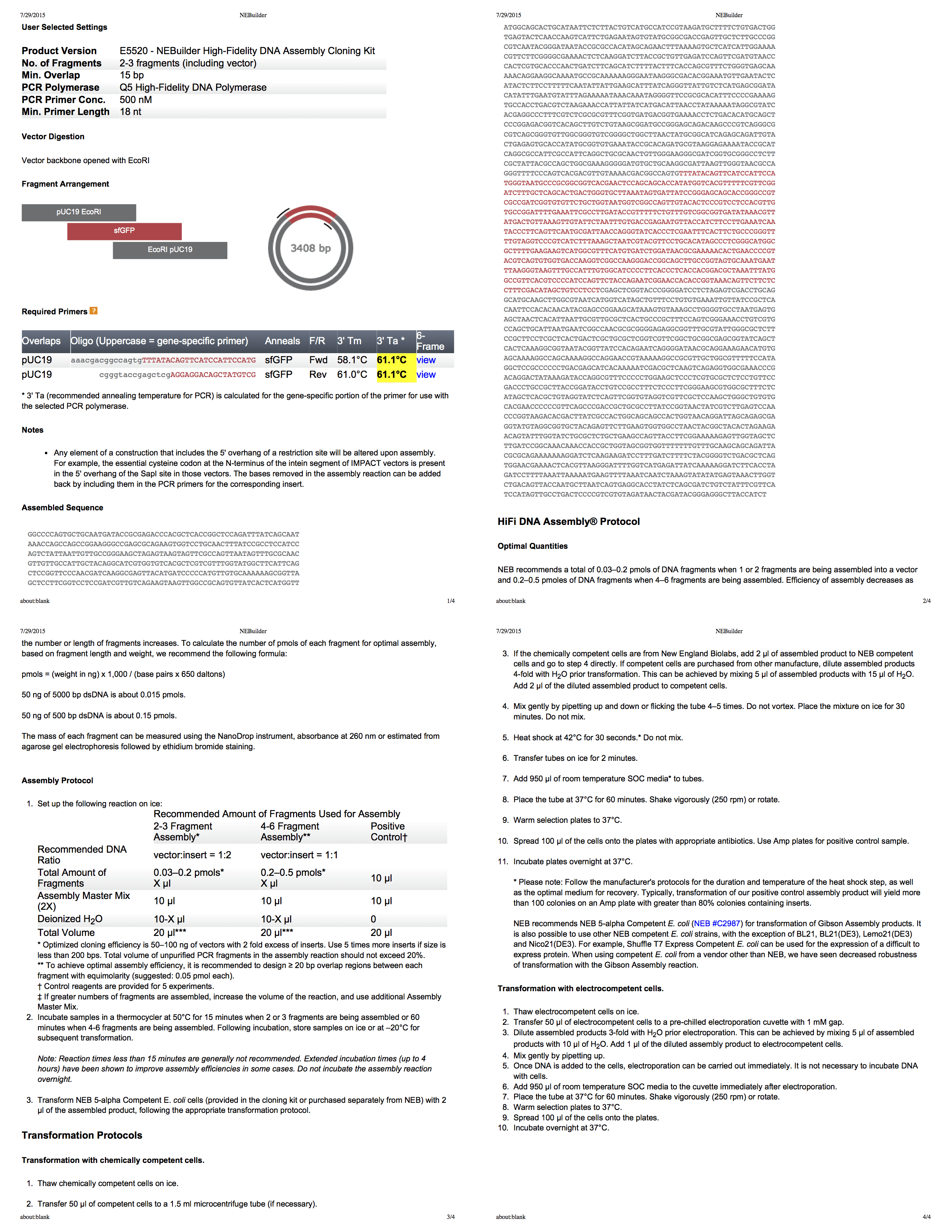
NEBuilder para ensamblaje Gibson
El NEBuilder de Biolab es una herramienta realmente excelente para crear el protocolo de compilación Gibson. Incluso le genera un PDF completo de cuatro páginas con toda la información. Usando esta herramienta, desarrollamos un protocolo para cortar pUC19 con EcoRI, y luego usamos PCR [PCR, la reacción en cadena de la polimerasa permite lograr un aumento significativo en pequeñas concentraciones de ciertos fragmentos de ADN en material biológico - aprox. por.] para agregar fragmentos del tamaño apropiado a la inserción.
Segunda parte: experimento
El experimento consta de cuatro etapas:
- Reacción de inserción de la cadena de polimerasa para agregar material con una secuencia de flanqueo.
- Cortar un plásmido para acomodar la inserción.
- Montaje por inserción de Gibson y plásmidos.
- Transformación de bacterias utilizando el plásmido ensamblado.
Paso 1. Inserción de PCR
El ensamblaje de Gibson depende de la secuencia de ADN que recolecte, que tenga una secuencia superpuesta (consulte el protocolo NEB con instrucciones detalladas más arriba). Además de la amplificación simple, la PCR también le permite agregar una secuencia de ADN flanqueante simplemente incluyendo una secuencia adicional en los cebadores (también se puede clonar
usando solo OE-PCR ).
Sintetizamos cebadores de acuerdo con el protocolo NEB anterior.
Probé el protocolo Quickstart en el sitio Transcriptic, pero todavía hay
un comando de protocolo automático . Transcriptic en sí no sintetiza oligonucleótidos, por lo que después de 1-2 días de espera, estos cebadores aparecen mágicamente en mi inventario (tenga en cuenta que la parte específica del gen de los cebadores se indica en mayúscula a continuación, pero estas son solo cosas cosméticas).
insert_primers = ["aaacgacggccagtgTTTATACAGTTCATCCATTCCATG", "cgggtaccgagctcgAGGAGGACAGCTATGTCG"]
Análisis de cebadores
Puede analizar las propiedades de estos cebadores utilizando el
IDT OligoAnalyzer . PCR
primer dimer , NEB .
Gene-specific portion of flank (uppercase)
Melt temperature: 51C, 53.5C
Full sequence
Melt temperature: 64.5C, 68.5C
Hairpin: -.4dG, -5dG
Self-dimer: -9dG, -16dG
Heterodimer: -6dG
PCR, , PCR. ( ), : . . , — .
Código """ PCR overlap extension of sfGFP according to NEB protocol. v5: Use 3/10ths as much primer as the v4 protocol. v6: more complex touchdown pcr procedure. The Q5 temperature was probably too hot v7: more time at low temperature to allow gene-specific part to anneal v8: correct dNTP concentration, real touchdown """ p = Protocol()
WARNING:root:Low volume for well sfGFP 1 /sfGFP 1 : 2.0:microliter
sfGFP 1 /sfGFP 1 2.0:microliter {'dilution': '0.25ng/ul'}
sfgfp_pcroe_v5_puc19_primer1_10uM 75.0:microliter {}
sfgfp_pcroe_v5_puc19_primer2_10uM 75.0:microliter {}
Consolidated volume 52.0:microliter
Protocol 1. Amplify the insert (oligos previously synthesized)
---------------------------------------------------------------
✓ Protocol analyzed
11 instructions
8 containers
Total Cost: $32.18
Workcell Time: $4.32
Reagents & Consumables: $27.86 : PCR
( )
( ). , , .
D1, E1, F1 2 , 4 8 . (50 ). , .
GelEval , , , , . . GelEval 40 /.
,
, dNTP , , 12,5 , 6 740bp 25 . GelEval 40 x 25 (1 2 ), , .
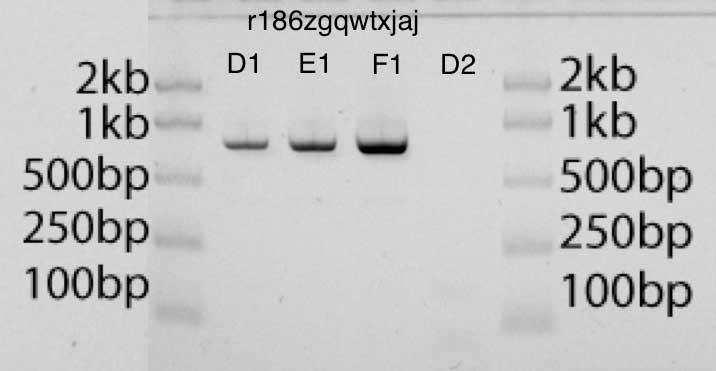 - EcoRI- pUC19, (D1, E1, F1), (D2)
- EcoRI- pUC19, (D1, E1, F1), (D2)PCR
Transcriptic . , .
, . 35 PCR,
PCR . — ! — , PCR , .
 PCR: , 35 42
PCR: , 35 422.
Para insertar nuestro ADN sfGFP en pUC19, primero debe cortar el plásmido. Siguiendo el protocolo NEB, hago esto usando la enzima de restricción EcoRI . En el inventario transcriptico estándar hay reactivos que necesito: estos son NEB EcoRI y 10x CutSmart buffer , así como el plásmido NEB pUC19 .Para información, a continuación se encuentran los precios de su inventario. De hecho, pago solo una parte del precio, ya que Transcriptic toma el pago por la cantidad realmente consumida: ID del artículo Cantidad Precio de concentración
------------ ------ ------------- ----------------- - ----
CutSmart 10x B7204S 5 ml 10 X $ 19.00
EcoRI R3101L 50,000 unidades 20,000 unidades / ml $ 225.00
pUC19 N3041L 250 μg 1,000 μg / ml $ 268.00
Seguí el protocolo NEB tanto como pude:. 10X dH2O 1X. , , , , . 50 5 10x NEBuffer , dH2O.
, 1 λ 1 37°C 50 . , 5-10 10-20 1- .
1 50 .
Código """Protocol for cutting pUC19 with EcoRI.""" p = Protocol() experiment_name = "puc19_ecori_v3" options = {} inv = { 'water': "rs17gmh5wafm5p",
Volumes: re_tube:135.0:microliter water_tube:383.0:microliter EcoRI:30.0:microliter
Consolidated volume: 78.0:microliter
✓ Protocol analyzed
12 instructions
5 containers
Total Cost: $30.72
Workcell Time: $3.38
Reagents & Consumables: $27.34
:
, . .
«» ( 1,5 15 !). , D1 E1 ( ). , EcoRI .
, D1 E1 2,6kb. D2 : , .
-. , Transcriptic .
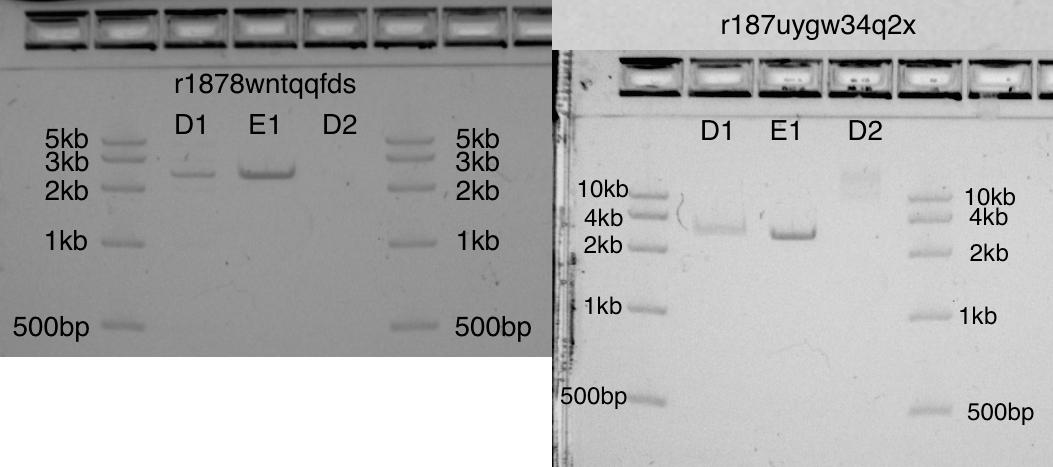 , pUC19 (2,6kb) D1 E1, pUC19 D2
, pUC19 (2,6kb) D1 E1, pUC19 D23.
, — ,
M13 ( )
qPCR , , . , , , .
, M13 , M13.
Código """Debugging transformation protocol: Gibson assembly followed by qPCR and a gel v2: include v3 Gibson assembly""" p = Protocol() options = {} experiment_name = "debug_sfgfp_puc19_gibson_seq_v2" inv = { "water" : "rs17gmh5wafm5p",
WARNING:root:Low volume for well sfgfp_puc19_gibson_v1_clone/sfgfp_puc19_gibson_v1_clone : 11.0:microliter
✓ Protocol analyzed
11 instructions
6 containers
Total Cost: $32.09
Tiempo de celda de trabajo: $ 6.98
Reactivos y consumibles: $ 25.11
Resultados: qPCR para ensamblaje Gibson
Puedo acceder a los datos de qPCR en formato JSON a través de la API transcriptica. Esta característica no está bien documentada , pero puede ser extremadamente útil. Las API incluso le dan acceso a algunos datos de diagnóstico de robots, que pueden ayudar con la depuración.Primero, solicitamos datos de lanzamiento: project_id, run_id = "p16x6gna8f5e9", "r18mj3cz3fku7" api_url = "https://secure.transcriptic.com/hgbrian/{}/runs/{}/data.json".format(project_id, run_id) data_response = requests.get(api_url, headers=tsc_headers) data = data_response.json()
Luego, especificamos esta identificación para obtener los datos de "procesamiento posterior" de qPCR: qpcr_id = data['debug_sfgfp_puc19_gibson_seq_v1_qpcr']['id'] pp_api_url = "https://secure.transcriptic.com/data/{}.json?key=postprocessed_data".format(qpcr_id) data_response = requests.get(pp_api_url, headers=tsc_headers) pp_data = data_response.json()
Aquí están los valores de Ct (umbral de ciclo) para cada tubo. Ct es simplemente el punto en el que la fluorescencia excede un cierto valor. Ella dice aproximadamente cuánto ADN hay en este momento (y, por lo tanto, aproximadamente dónde comenzamos).
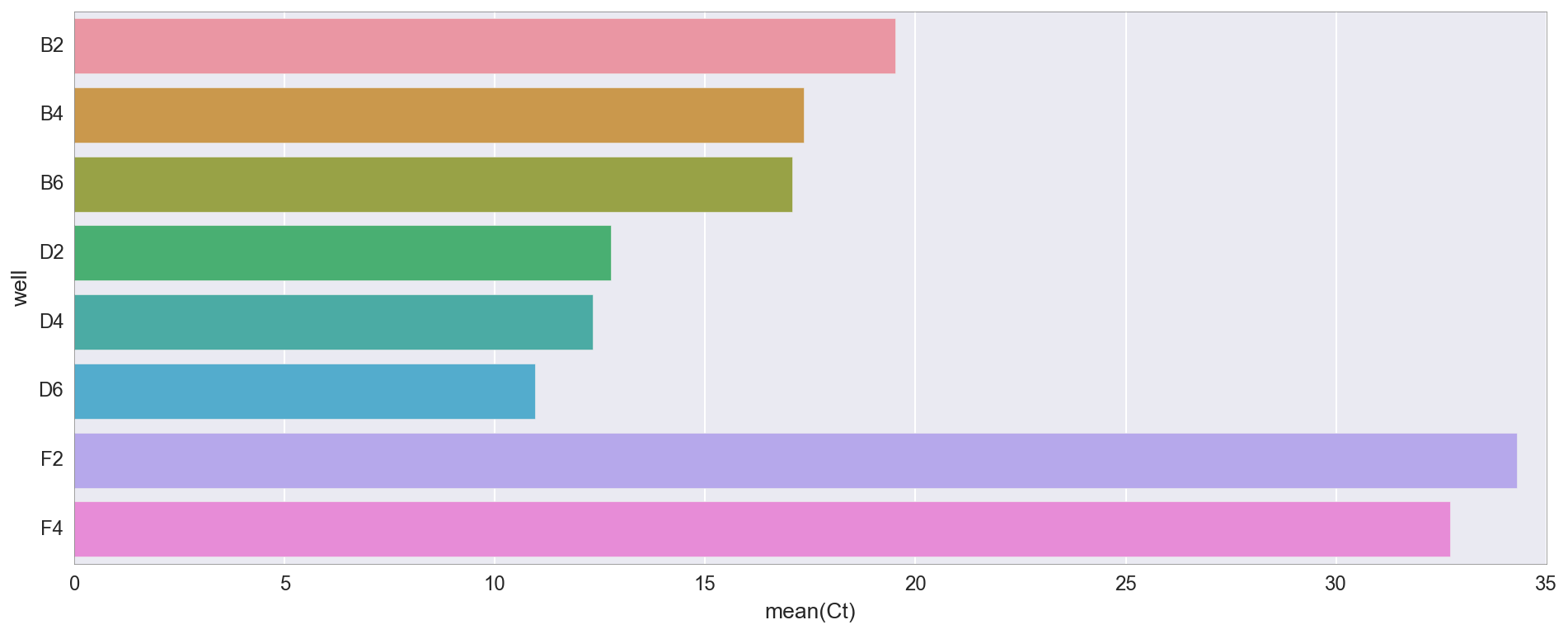
, D2/4/6 ( «v3»), B2/4/6 ( «v1»). v1 v3 , v3 4X NEB, . 30 (F2, F4), -, , .
qPCR, .
f, ax = plt.subplots(figsize=(16,6)) ax.set_color_cycle(['#fb6a4a', '#de2d26', '#a50f15', '#74c476', '#31a354', '#006d2c', '#08519c', '#6baed6']) amp0 = pp_data['amp0']['SYBR']['baseline_subtracted'] _ = [plt.plot(amp0[w_n[well]], label=well) for well in ['B2', 'B4', 'B6', 'D2', 'D4', 'D6', 'F2', 'F4']] _ = ax.set_ylim(0,) _ = plt.title("qPCR (reds=Gibson v1, greens=Gibson v3, blues=control)") _ = plt.legend(bbox_to_anchor=(1, .75), bbox_transform=plt.gcf().transFigure)
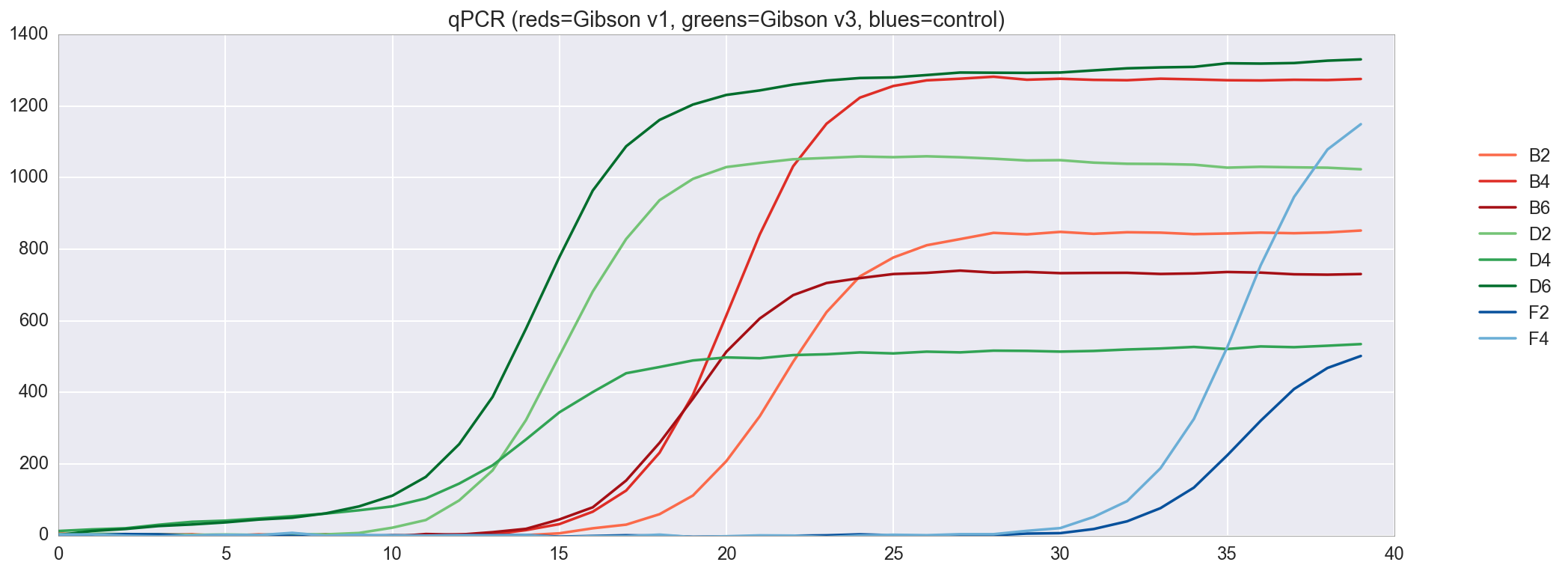
, qPCR , . v3 , v1, .
:
, 1kb B2, B4, B6, D2, D4, D6: ( 740bp, M13 — 40bp ). . , F2 F4 .
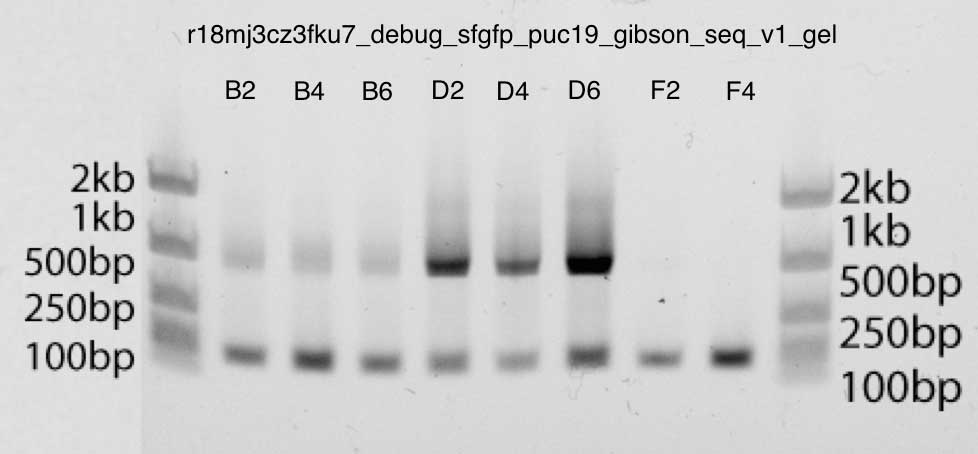 : v3 (D2, D4, D6), qPCR
: v3 (D2, D4, D6), qPCR4.
— .
E. coli sfGFP- pUC19.
Zymo DH5α Mix&Go . — Transcriptic. , , , , . , , .
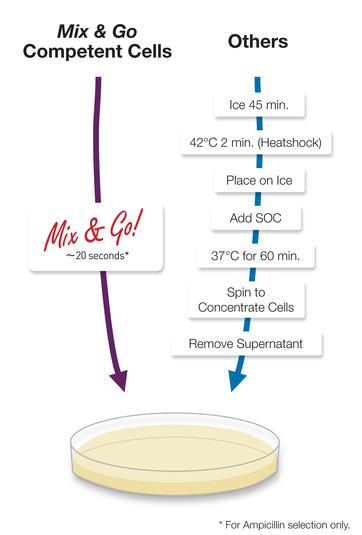 Zymo Mix & Go
Zymo Mix & Go— , . (« »), , («, »). , .
. , 37°C. , , , , Transcriptic — , . , , - , . . .
: (, , Mix&Go ); (, ); (, PCR ).
, , , , . , , !
, , , pUC19 (. . sfGFP) . pUC19 , , .
(«6-flat» Transcriptic), , . , , , . .
Código """Simple transformation protocol: transformation with unaltered pUC19""" p = Protocol() experiment_name = "debug_sfgfp_puc19_gibson_v1" inv = { "water" : "rs17gmh5wafm5p",
✓ Protocol analyzed
43 instructions
3 containers
$45.43
:
, ( ) , , . , Transcriptic , .
( ) , . , , , , 55 10 . . , . , , .
( , , ,
E. coli . El crecimiento es mucho más débil en las placas de ampicilina, aunque hay muchas más bacterias allí, como se esperaba).En general, la transformación funcionó lo suficientemente bien como para continuar, aunque hay algunos defectos. Placas de células transformadas con pUC19 después de 18 horas: sin antibiótico (izquierda) y con antibiótico (derecha)
Placas de células transformadas con pUC19 después de 18 horas: sin antibiótico (izquierda) y con antibiótico (derecha)Transformación del producto después del montaje.
Dado que el ensamblaje Gibson y la simple transformación pUC19 parecen funcionar, ahora puede probar la transformación con un plásmido completamente ensamblado que exprese sfGFP.Además de la inserción recopilada, también agregaré un poco de IPTG y X-gal a las placas para ver la conversión exitosa utilizando el método de selección azul y blanco . Esta información adicional es útil, porque si la transformación se lleva a cabo con el pUC19 habitual, que no contiene sfGFP, todavía dará resistencia a los antibióticos.Absorción y fluorescencia.
Según esta tabla , sfGFP brilla mejor a longitudes de onda de excitación de 485 nm / 510 nm. Encontré que en Transcriptic, 485/535 funciona mejor. Supongo que porque 485 y 510 son demasiado similares. Mido el crecimiento bacteriano a 600 nm ( OD600 ).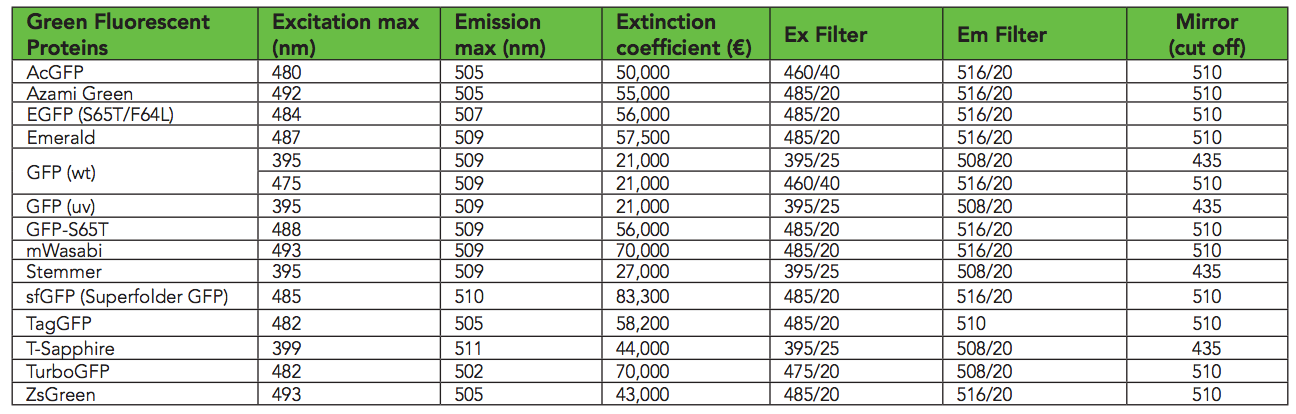 Variedad de GFP ( biotek )
Variedad de GFP ( biotek )IPTG y X-gal
IPTG 1M 1:1000. , X-gal 20 / 1:1000 (20 /). , 2000µl LB 2 .
40 X-gal 20 / 40 IPTG 0,1 mM ( 4 IPTG 1M), 30 . , IPTG, X-gal .
Código """Full Gibson assembly and transformation protocol for sfGFP and pUC19 v1: Spread IPTG and X-gal onto plates, then spread cells v2: Mix IPTG, X-gal and cells; spread the mixture v3: exclude X-gal so I can do colony picking better v4: repeat v3 to try other excitation/emission wavelengths""" p = Protocol() options = { "gibson" : False,
Inventory: IPTG/IPTG/IPTG/IPTG/IPTG/IPTG 832.0:microliter {}
Inventory: sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone 57.0:microliter {}
✓ Protocol analyzed
40 instructions
8 containers
Total Cost: $53.20
Workcell Time: $17.35
Reagents & Consumables: $35.86 , «» 96- . (
autopick ).
Código """Pick colonies from plates and grow in amp media and check for fluorescence. v2: try again with a new plate (no blue colonies) v3: repeat with different emission and excitation wavelengths""" p = Protocol() options = {} for k, v in list(options.items()): if v is False: del options[k] experiment_name = "sfgfp_puc19_gibson_pick_v3" def plate_expid(val): """refer to the previous plating experiment's outputs""" plate_exp = "sfgfp_puc19_gibson_plates_v4" return "{}_{}".format(plate_exp, val)
✓ Protocol analyzed
62 instructions
8 containers
Total Cost: $66.38
Workcell Time: $57.59
Reagents & Consumables: $8.78
:
– , , (1-4) (5-6). , , , , IPTG X-gal, Transcriptic.
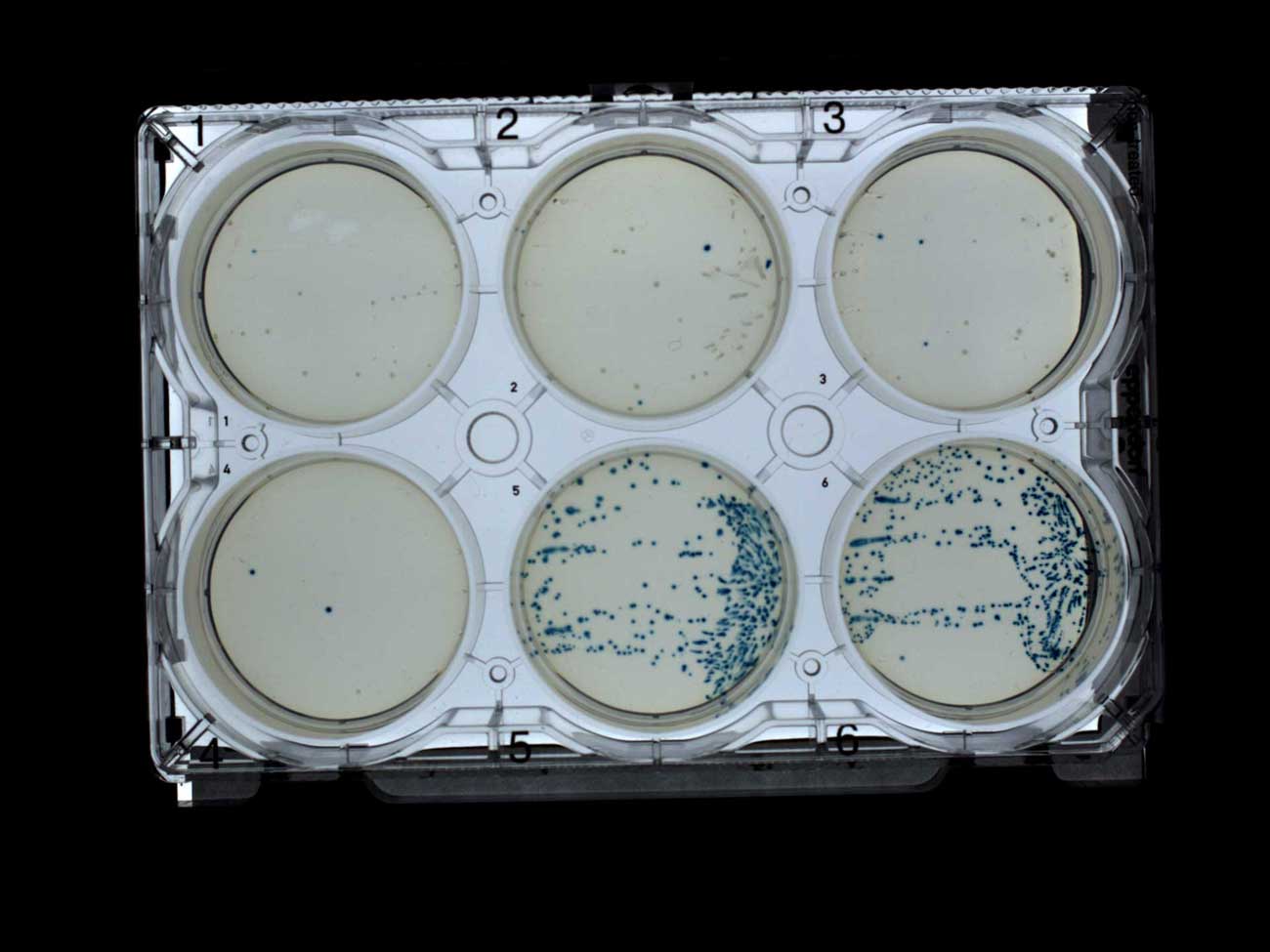 - (1-4) (5-6)
- (1-4) (5-6)- - . (
GraphicsMagick ). , , ( , ).
, Transcriptic. , 10 . , , . . , , , , .
 - (1-4) (5-6),El cribado azul y blanco tuvo un propósito específico. Mostró que la mayoría de las colonias se transforman correctamente. Al menos hay una inserción. Sin embargo, para una mejor colección de colonias, repetí el experimento sin X-gal.Solo con colonias blancas el robot recolector ensambló con éxito diez colonias de cada una de las primeras cinco placas. Se puede suponer que en la mayoría de las colonias recolectadas hay inserciones exitosas.
- (1-4) (5-6),El cribado azul y blanco tuvo un propósito específico. Mostró que la mayoría de las colonias se transforman correctamente. Al menos hay una inserción. Sin embargo, para una mejor colección de colonias, repetí el experimento sin X-gal.Solo con colonias blancas el robot recolector ensambló con éxito diez colonias de cada una de las primeras cinco placas. Se puede suponer que en la mayoría de las colonias recolectadas hay inserciones exitosas. Colonias que crecen en placas con ampicilina (1-4) y sin antibiótico (5-6)
Colonias que crecen en placas con ampicilina (1-4) y sin antibiótico (5-6)Resultados: transformación con producto ensamblado
Después de cultivar 50 colonias seleccionadas en una placa de 96 tubos durante 20 horas, mido la fluorescencia para verificar la expresión de sfGFP. Transcriptic utiliza un lector Tecan Infinite para medir la fluorescencia y la absorción (y la luminiscencia, si lo desea) ., , , sfGFP. , , - , sfGFP . , sfGFP, , , , .
(OD600) 20 ( 60 ).
for t in [0,4,8,12,16,20]: abs_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_abs_{}.csv".format(t), index_col="Well") flr_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_fl2_{}.csv".format(t), index_col="Well") if t == 0: new_data = abs_data.join(flr_data) else: new_data = new_data.join(abs_data, rsuffix='_{}'.format(t)) new_data = new_data.join(flr_data, rsuffix='_{}'.format(t)) new_data.columns = ["OD 600:nanometer_0", "Fluorescence_0"] + list(new_data.columns[2:])
Colocamos en el gráfico los datos de la hora 20 y los rastros de mediciones anteriores. De hecho, solo me interesan los últimos datos, ya que es entonces cuando se debe observar un pico de fluorescencia. svg = [] W, H = 800, 500 min_x, max_x = 0, 0.8 min_y, max_y = 0, 50000 def _toxy(x, y): return W*(x-min_x)/(max_x-min_x), HH*(y-min_y)/(max_y-min_y) def _topt(x, y): return ','.join(map(str,_toxy(x,y))) ab_fls = [[row[0]] + [list(row[1])] for row in new_data.iterrows()]
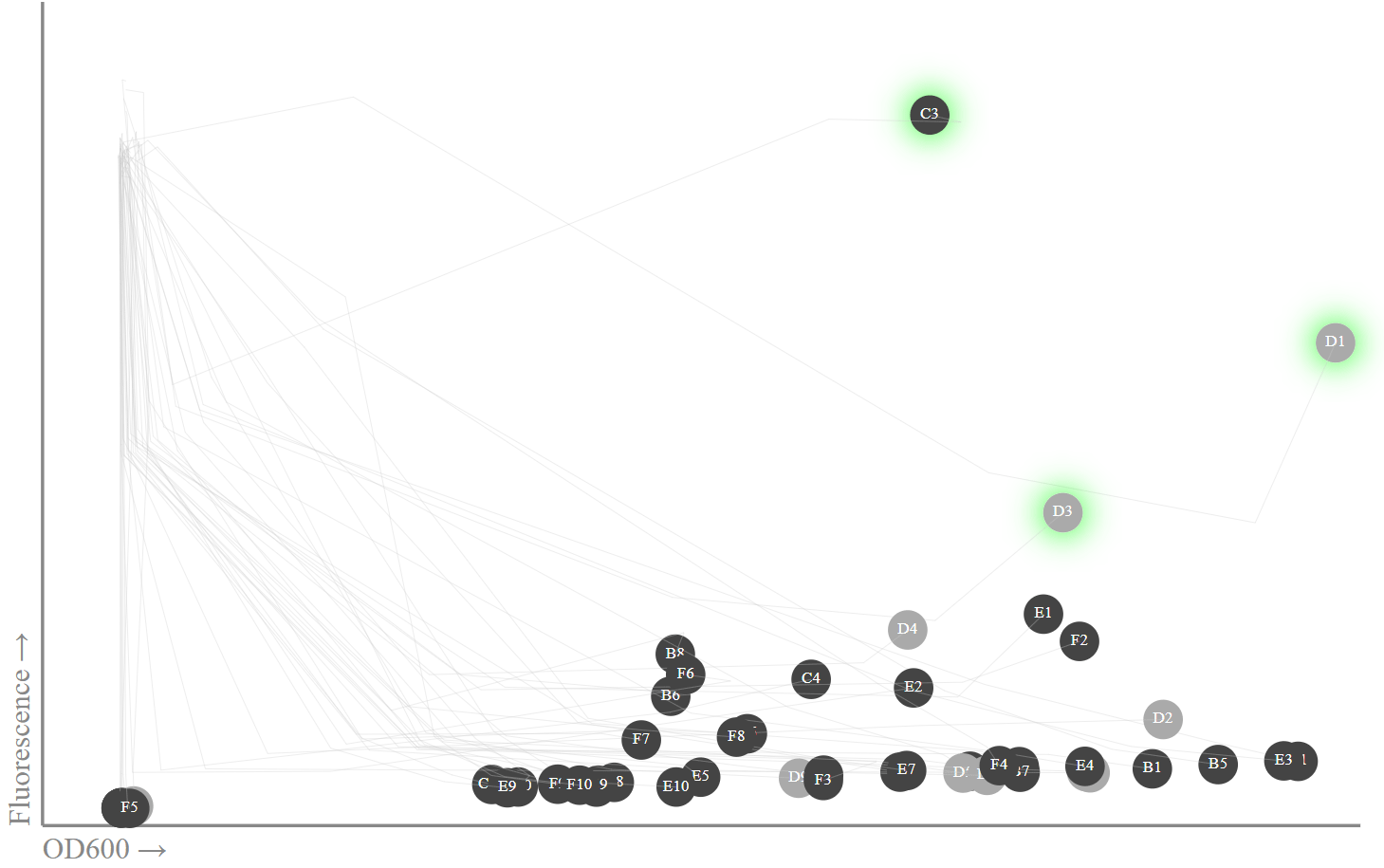 OD600: , . , sfGFPminiprep
OD600: , . , sfGFPminiprep , , 13. , - miniprep - Transcriptic, . (C1, D1, D3) (B1, B3, E1), sfGFP
muscle .
C1, D3 D3 sfGFP, B1, B3 E1 .
, . , 0 (40 000 ). 20- OD600 (, - ), . , , , , 11-15 .
(. . , ), , , ).
, , 50 sfGFP . , . , ( , , miniprep), 200 , .
, . , GFP, Python!
:
Precio
, , $360, :
- $70
- $32 PCR
- $31
- $32
- $53
- $67
- $75 3 miniprep'
, $250-300 . , 50 , , .
, ( ) ( IT). Transcriptic , . , , . , , , , .
, . , - , . , : , , IDT .
:
, . , :
- ! , . autoprotocol, .
- . 100 , .
- , , PCR. , , ? / ? , , , « 2-3 ». ?
- . . , .
- . .
- . . , 1 96 (96−x) 96- , .
- . csv , .
- . - , .
, , , . , 1994 :
- Transcriptic — . , , , . , , .
- — Transcriptic.
- , . Transcriptic ( , , ).
- , ( : ~$0). , .
- Transcriptic . , , .
, , - , , .
, :
- Twist/IDT/Gen9 Transcriptic (, - ).
- , , , , . .
- ( NEB, IDT) (, primer3 ).
(
) , ,
. , in vivo (. . ).
, , : RBS , ; ; .
?
, . :
- - //, .
- , , .
- in vivo split-GFP .
- scFv . scFvs - .
- BiTE , ( , ).
- Haga una vacuna local que ingrese al cuerpo a través de los folículos pilosos (no recomiendo probar esto en casa).
- Mutagenice su proteína de cientos de maneras diferentes y vea qué sucede. ¿Entonces escalar a 1000 o 10,000 mutaciones? ¿Quizás caracterizar las mutaciones de GFP?
Para obtener nuevas ideas sobre lo que es posible con el diseño de proteínas, mire cientos de proyectos de iGEM .Al final, quiero agradecer al Transcriptic Ben Miles por su ayuda para completar este proyecto.