Cómo leer este artículo : Pido disculpas por el hecho de que el texto resultó tan largo y caótico. Para ahorrarle tiempo, comienzo cada capítulo con la introducción de "Lo que aprendí", en el que explico la esencia del capítulo en una o dos oraciones.
"¡Solo muestra la solución!" Si solo quiere ver a qué he venido, vaya al capítulo "Sea más inventivo", pero creo que es más interesante y útil leer sobre los fracasos.Recientemente, me dieron instrucciones de configurar un proceso para procesar un gran volumen de las secuencias de ADN originales (técnicamente, este es un chip SNP). Era necesario obtener rápidamente datos sobre una ubicación genética dada (llamada SNP) para el modelado posterior y otras tareas. Con la ayuda de R y AWK, pude limpiar y organizar los datos de forma natural, acelerando enormemente el procesamiento de las solicitudes. Esto no fue fácil para mí y requirió numerosas iteraciones. Este artículo te ayudará a evitar algunos de mis errores y a demostrar lo que hice al final.
Primero, algunas explicaciones introductorias.
Datos
Nuestro Centro de Procesamiento de Información Genética de la Universidad nos ha proporcionado 25 TB de datos TSV. Los dividí en 5 paquetes comprimidos por Gzip, cada uno de los cuales contenía unos 240 archivos de cuatro gigabytes. Cada fila contenía datos para un SNP de una persona. En total, se transmitieron datos sobre ~ 2.5 millones de SNP y ~ 60 mil personas. Además de la información de SNP, había numerosas columnas en los archivos con números que reflejaban diversas características, como la intensidad de lectura, la frecuencia de diferentes alelos, etc. Había alrededor de 30 columnas con valores únicos.
Propósito
Al igual que con cualquier proyecto de gestión de datos, lo más importante era determinar cómo se utilizarían los datos. En este caso,
en su mayor parte, seleccionaremos modelos y flujos de trabajo para SNP basados en SNP . Es decir, al mismo tiempo necesitaremos datos para un solo SNP. Tuve que aprender a extraer todos los registros relacionados con uno de los 2.5 millones de SNP de la manera más simple, rápida y económica.
Como no hacerlo
Citaré un cliché adecuado:
No fallé mil veces, solo descubrí mil maneras de no analizar un montón de datos en un formato conveniente para consultas.
Primer intento
Lo que aprendí : no hay una forma barata de analizar 25 TB a la vez.
Después de escuchar el tema "Métodos avanzados de procesamiento de Big Data" en la Universidad de Vanderbilt, estaba seguro de que era un sombrero. Quizás llevará una o dos horas configurar el servidor Hive para que ejecute todos los datos e informe sobre el resultado. Dado que nuestros datos se almacenan en AWS S3, utilicé el servicio
Athena , que le permite aplicar consultas Hive SQL a los datos S3. No es necesario configurar / elevar el clúster Hive, e incluso pagar solo por los datos que está buscando.
Después de mostrarle a Athena mis datos y su formato, realicé algunas pruebas con consultas similares:
select * from intensityData limit 10;
Y rápidamente obtuvo resultados bien estructurados. Listo
Hasta que intentamos usar los datos en el trabajo ...
Me pidieron que extrajera toda la información de SNP para probar el modelo. Ejecuté una consulta:
select * from intensityData where snp = 'rs123456';
... y esperé. Después de ocho minutos y más de 4 TB de los datos solicitados, obtuve el resultado. Athena cobra una tarifa por la cantidad de datos encontrados, a $ 5 por terabyte. Entonces, esta única solicitud cuesta $ 20 y ocho minutos de espera. Para ejecutar el modelo de acuerdo con todos los datos, era necesario esperar 38 años y pagar $ 50 millones. Obviamente, esto no nos convenía.
Era necesario usar Parquet ...
Lo que aprendí : tenga cuidado con el tamaño de sus archivos de Parquet y su organización.
Al principio traté de corregir la situación convirtiendo todos los TSV en
archivos de Parquet . Son convenientes para trabajar con grandes conjuntos de datos, porque la información en ellos se almacena en forma de columnas: cada columna se encuentra en su propio segmento de memoria / disco, a diferencia de los archivos de texto en los que las líneas contienen elementos de cada columna. Y si necesita encontrar algo, simplemente lea la columna necesaria. Además, se almacena un rango de valores en cada archivo en una columna, por lo que si el valor deseado no está en el rango de la columna, Spark no perderá tiempo escaneando todo el archivo.
Ejecuté una simple tarea de
AWS Glue para convertir nuestros TSV a Parquet y solté nuevos archivos en Athena. Tomó alrededor de 5 horas. Pero cuando lancé la solicitud, me llevó casi el mismo tiempo y un poco menos de dinero completarla. El hecho es que Spark, tratando de optimizar la tarea, simplemente desempacó un trozo de TSV y lo colocó en su propio trozo de Parquet. Y dado que cada fragmento era lo suficientemente grande y contenía los registros completos de muchas personas, todos los SNP se almacenaban en cada archivo, por lo que Spark tuvo que abrir todos los archivos para extraer la información necesaria.
Curiosamente, el tipo de compresión predeterminado (y recomendado) en Parquet (rápido) no es divisible. Por lo tanto, cada ejecutor se aferró a la tarea de desempaquetar y descargar el conjunto de datos completo de 3.5 GB.

Entendemos el problema
Lo que aprendí : la clasificación es difícil, especialmente si los datos se distribuyen.
Me pareció que ahora entendía la esencia del problema. Todo lo que tenía que hacer era ordenar los datos por columna SNP, no por personas. Luego, se almacenarán varios SNP en una porción de datos separada, y luego la función inteligente Parquet "abrirá solo si el valor está en el rango" se manifestará en todo su esplendor. Desafortunadamente, clasificar miles de millones de filas dispersas en un clúster ha demostrado ser una tarea desalentadora.
AWS ciertamente no quiere devolver el dinero porque "soy un estudiante distraído". Después de que comencé a ordenar en Amazon Glue, funcionó durante 2 días y se bloqueó.
¿Qué pasa con la partición?
Lo que aprendí : las particiones en Spark deben estar equilibradas.
Entonces se me ocurrió la idea de particionar los datos en los cromosomas. Hay 23 de ellos (y algunos más, dado el ADN mitocondrial y las áreas no mapeadas).
Esto le permitirá dividir los datos en porciones más pequeñas. Si agrega una sola línea de
partition_by = "chr" a la función de exportación de Spark en la secuencia de comandos de Glue, los datos deben clasificarse en cubos.
 El genoma consta de numerosos fragmentos llamados cromosomas.
El genoma consta de numerosos fragmentos llamados cromosomas.Lamentablemente, esto no funcionó. Los cromosomas tienen diferentes tamaños y, por lo tanto, una cantidad diferente de información. Esto significa que las tareas que Spark envió a los trabajadores no estaban equilibradas y se realizaban lentamente, porque algunos nodos finalizaron antes y estaban inactivos. Sin embargo, las tareas se completaron. Pero al solicitar un SNP, el desequilibrio nuevamente causó problemas. El costo de procesar SNP en cromosomas más grandes (es decir, de dónde queremos obtener los datos) disminuyó solo unas 10 veces. Mucho, pero no lo suficiente.
¿Y si te divides en particiones aún más pequeñas?
Lo que aprendí : nunca intente hacer 2.5 millones de particiones.
Decidí dar un paseo y particioné cada SNP. Esto garantizó el mismo tamaño de particiones.
Mal fue una idea . Aproveché Glue y agregué la inocente
partition_by = 'snp' . La tarea comenzó y comenzó a ejecutarse. Un día después, verifiqué y vi que hasta ahora no se había escrito nada en S3, así que eliminé la tarea. Parece que Glue estaba escribiendo archivos intermedios en un lugar oculto en S3, y muchos archivos, tal vez un par de millones. Como resultado, mi error costó más de mil dólares y no agradó a mi mentor.
Particionamiento + clasificación
Lo que aprendí : la clasificación sigue siendo difícil, como lo es configurar Spark.
El último intento de particionamiento fue que particioné los cromosomas y luego clasifiqué cada partición. En teoría, esto aceleraría cada solicitud, porque los datos SNP deseados deberían estar dentro de varios fragmentos de Parquet dentro de un rango determinado. Por desgracia, ordenar incluso datos particionados ha resultado ser una tarea difícil. Como resultado, cambié a EMR para un clúster personalizado y usé ocho instancias potentes (C5.4xl) y Sparklyr para crear un flujo de trabajo más flexible ...
# Sparklyr snippet to partition by chr and sort w/in partition # Join the raw data with the snp bins raw_data group_by(chr) %>% arrange(Position) %>% Spark_write_Parquet( path = DUMP_LOC, mode = 'overwrite', partition_by = c('chr') )
... sin embargo, la tarea aún no se completó. Me sintonicé en todos los sentidos: aumenté la asignación de memoria para cada ejecutor de consultas, usé nodos con una gran cantidad de memoria, utilicé variables de transmisión, pero cada vez resultó ser medias tintas, y gradualmente los artistas comenzaron a fallar, hasta que todo se detuvo.
Me estoy volviendo más inventivo
Lo que aprendí : a veces los datos especiales requieren soluciones especiales.
Cada SNP tiene un valor de posición. Este es el número correspondiente al número de bases que se encuentran a lo largo de su cromosoma. Esta es una forma buena y natural de organizar nuestros datos. Al principio quería particionar por región de cada cromosoma. Por ejemplo, puestos 1 - 2000, 2001 - 4000, etc. Pero el problema es que los SNP no se distribuyen uniformemente entre los cromosomas, por lo que el tamaño de los grupos variará enormemente.
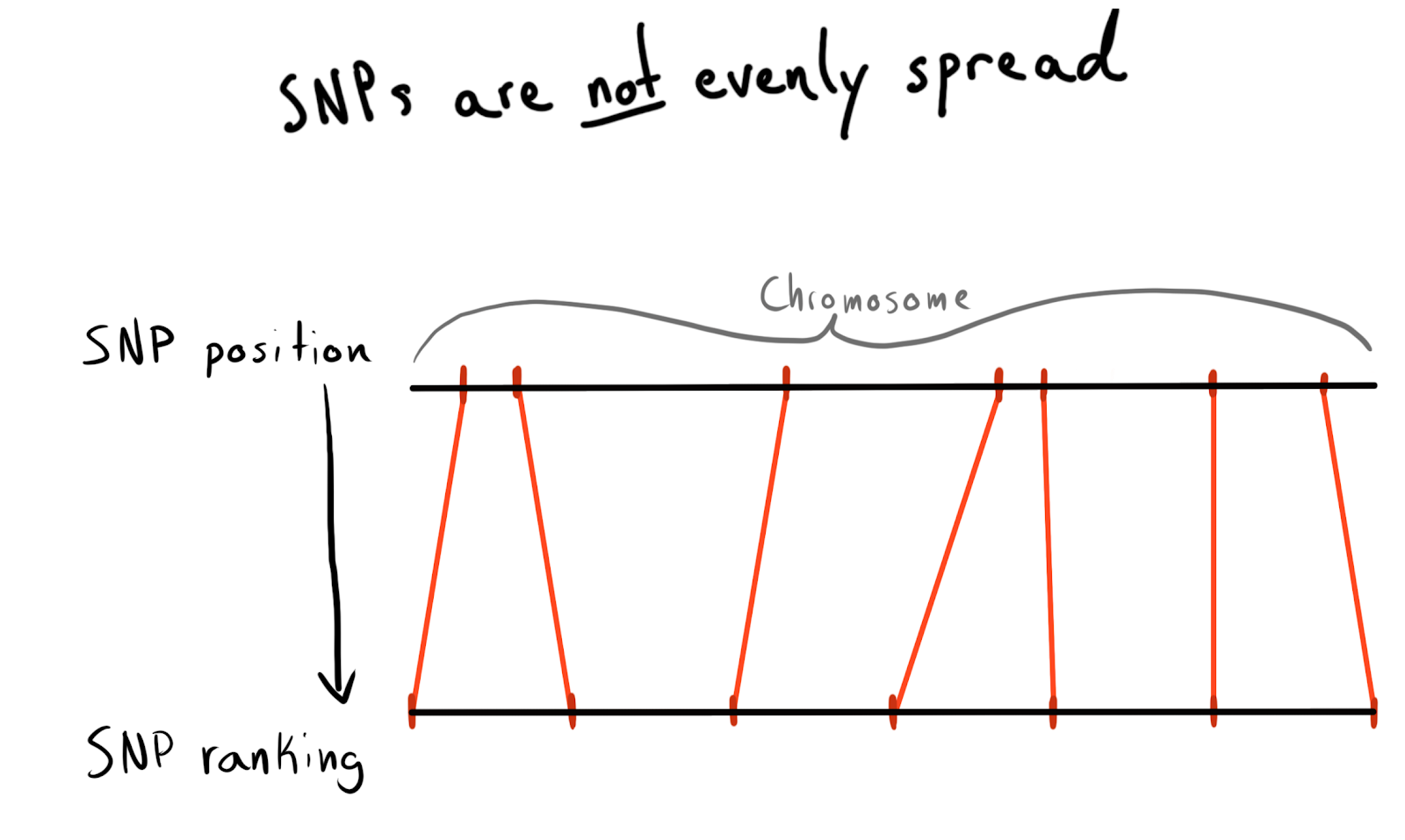
Como resultado, llegué a dividirme en posiciones de categorías (rango). Según los datos ya descargados, solicité una lista de SNP únicos, sus posiciones y cromosomas. Luego clasificó los datos dentro de cada cromosoma y recolectó SNP en grupos (bin) de un tamaño dado. Diga 1000 SNP cada uno. Esto me dio una relación SNP con un grupo en cromosoma.
Al final, hice grupos (bin) en 75 SNP, explicaré la razón a continuación.
snp_to_bin <- unique_snps %>% group_by(chr) %>% arrange(position) %>% mutate( rank = 1:n() bin = floor(rank/snps_per_bin) ) %>% ungroup()
Primero prueba con Spark
Lo que aprendí : la integración de Spark es rápida, pero la partición sigue siendo costosa.
Quería leer este pequeño marco de datos (2,5 millones de líneas) en Spark, combinarlo con datos sin procesar y luego particionar por la columna
bin recién agregada.
sdf_broadcast() , por lo que Spark descubre que debería enviar un marco de datos a todos los nodos. Esto es útil si los datos son pequeños y necesarios para todas las tareas. De lo contrario, Spark intenta ser inteligente y distribuye los datos según sea necesario, lo que puede causar frenos.
Y nuevamente, mi idea no funcionó: las tareas funcionaron por un tiempo, completaron la fusión y luego, como los ejecutores iniciados por la partición, comenzaron a fallar.
Añadir AWK
Lo que aprendí : no duermas cuando lo básico te enseña. Seguramente alguien ya resolvió su problema en la década de 1980.
Hasta este punto, la causa de todas mis fallas con Spark fue la confusión de datos en el clúster. Quizás la situación pueda mejorarse mediante el preprocesamiento. Decidí tratar de dividir los datos de texto sin procesar en columnas cromosómicas, por lo que esperaba proporcionar a Spark datos "pre-particionados".
Busqué en StackOverflow cómo desglosar los valores de columna y encontré
una gran respuesta. Con AWK, puede dividir un archivo de texto en valores de columna escribiendo en el script, en lugar de enviar los resultados a
stdout .
Para probar, escribí un script Bash. Descargué uno de los TSV empaquetados, luego lo descomprimí con
gzip y lo envié a
awk .
gzip -dc path/to/chunk/file.gz | awk -F '\t' \ '{print $1",..."$30">"chunked/"$chr"_chr"$15".csv"}'
Funcionó!
Relleno del núcleo
Lo que aprendí :
gnu parallel es algo mágico, todos deberían usarlo.
La separación fue bastante lenta, y cuando comencé a probar para usar una instancia EC2 potente (y costosa), resultó que estaba usando solo un núcleo y aproximadamente 200 MB de memoria. Para resolver el problema y no perder mucho dinero, fue necesario descubrir cómo paralelizar el trabajo. Afortunadamente, en el sorprendente
Data Science de Jeron Janssens
en el libro de la
Línea de Comando , encontré un capítulo sobre paralelización. De él aprendí sobre
gnu parallel , un método muy flexible para implementar multihilo en Unix.
Cuando comencé la partición usando un nuevo proceso, todo estaba bien, pero había un cuello de botella: la descarga de objetos S3 al disco no era demasiado rápida y no estaba completamente paralela. Para solucionar esto, hice esto:
- Descubrí que es posible implementar el paso de descarga S3 directamente en la tubería, eliminando por completo el almacenamiento intermedio en el disco. Esto significa que puedo evitar escribir datos en bruto en el disco y usar un almacenamiento aún más pequeño y, por lo tanto, más barato en AWS.
- El comando
aws configure set default.s3.max_concurrent_requests 50 aumentó en gran medida el número de subprocesos que utiliza la AWS CLI (hay 10 por defecto).
- Cambié a la instancia EC2 optimizada para la velocidad de la red, con la letra n en el nombre. Descubrí que la pérdida de potencia informática cuando se usan n-instancias está más que compensada por un aumento en la velocidad de descarga. Para la mayoría de las tareas, usé c5n.4xl.
- Cambié
gzip a pigz , esta es una herramienta gzip que puede hacer cosas geniales para paralelizar la tarea inicialmente incomparable de desempaquetar archivos (esto ayudó menos).
Estos pasos se combinan entre sí para que todo funcione muy rápidamente. Gracias a la mayor velocidad de descarga y al rechazo de la escritura en el disco, ahora podría procesar un paquete de 5 terabytes en solo unas pocas horas.
Se suponía que este tweet mencionaba 'TSV'. Por desgracia
Usar datos analizados nuevamente
Lo que aprendí : a Spark le encantan los datos sin comprimir y no le gusta combinar particiones.
Ahora los datos estaban en S3 en un formato desempaquetado (leído, compartido) y semi-ordenado, y pude volver a Spark nuevamente. Me esperaba una sorpresa: ¡otra vez no pude lograr lo deseado! Fue muy difícil decirle a Spark exactamente cómo se dividieron los datos. E incluso cuando hice esto, resultó que había demasiadas particiones (95 mil), y cuando reduje su número a límites coherentes con la
coalesce , arruinó mi partición. Estoy seguro de que esto se puede solucionar, pero en un par de días de búsqueda, no pude encontrar una solución. Al final, completé todas las tareas en Spark, aunque me llevó algo de tiempo, y mis archivos de Parquet divididos no eran muy pequeños (~ 200 Kb). Sin embargo, los datos estaban donde se necesitaban.
 Demasiado pequeño y diferente, maravilloso!
Demasiado pequeño y diferente, maravilloso!Probar solicitudes locales de Spark
Lo que aprendí : Spark tiene demasiados gastos generales para resolver problemas simples.
Al descargar los datos en un formato inteligente, pude probar la velocidad. Configuré un script en R para iniciar el servidor local de Spark, y luego cargué el marco de datos de Spark desde el repositorio especificado de los grupos de Parquet (bin). Traté de cargar todos los datos, pero no pude hacer que Sparklyr reconociera la partición.
sc <- Spark_connect(master = "local") desired_snp <- 'rs34771739' # Start a timer start_time <- Sys.time() # Load the desired bin into Spark intensity_data <- sc %>% Spark_read_Parquet( name = 'intensity_data', path = get_snp_location(desired_snp), memory = FALSE ) # Subset bin to snp and then collect to local test_subset <- intensity_data %>% filter(SNP_Name == desired_snp) %>% collect() print(Sys.time() - start_time)
La ejecución tomó 29.415 segundos. Mucho mejor, pero no demasiado bueno para probar algo en masa. Además, no pude acelerar el trabajo con el almacenamiento en caché, porque cuando intenté almacenar en caché el marco de datos en la memoria, Spark siempre se bloqueaba, incluso cuando asignaba más de 50 GB de memoria para un conjunto de datos que pesaba menos de 15.
Regresar a AWK
Lo que aprendí : los arrays asociativos de AWK son muy eficientes.
Comprendí que podía lograr una mayor velocidad. Recordé que en la excelente guía AWK de
Bruce Barnett , leí sobre una característica interesante llamada "
matrices asociativas ". De hecho, estos son pares clave-valor, que por alguna razón se llamaron de manera diferente en AWK y, por lo tanto, de alguna manera no los mencioné en particular.
Roman Cheplyaka recordó que el término "matrices asociativas" es mucho más antiguo que el término "par clave-valor". Incluso si
busca valor-clave en Google Ngram , no verá este término allí, ¡pero encontrará matrices asociativas! Además, el par clave-valor se asocia con mayor frecuencia a las bases de datos, por lo que es mucho más lógico compararlo con el hashmap. Me di cuenta de que podía usar estas matrices asociativas para conectar mis SNP a la tabla bin y los datos sin procesar sin usar Spark.
Para esto, en el script AWK, usé el bloque
BEGIN . Este es un fragmento de código que se ejecuta antes de que la primera línea de datos se transfiera al cuerpo principal del script.
join_data.awk BEGIN { FS=","; batch_num=substr(chunk,7,1); chunk_id=substr(chunk,15,2); while(getline < "snp_to_bin.csv") {bin[$1] = $2} } { print $0 > "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv" }
El comando
while(getline...) cargó todas las líneas del grupo CSV (bin), estableció la primera columna (nombre SNP) como la clave para la matriz asociativa
bin y el segundo valor (grupo) como el valor. Luego, en el bloque
{ } , que se aplica a todas las líneas del archivo principal, cada línea se envía al archivo de salida, que obtiene un nombre único según su grupo (bin):
..._bin_"bin[$1]"_...Las
chunk_id batch_num y
chunk_id correspondieron a los datos proporcionados por la canalización, lo que evitó el estado de la carrera, y cada hilo de ejecución lanzado por
parallel escribió en su propio archivo único.
Dado que dispersé todos los datos sin procesar en carpetas en los cromosomas que quedaron después de mi experimento anterior con AWK, ahora podría escribir otro script Bash para procesar en el cromosoma a la vez y dar datos particionados más profundos a S3.
DESIRED_CHR='13'
El guión tiene dos secciones
parallel .
La primera sección lee los datos de todos los archivos que contienen información sobre el cromosoma deseado, luego estos datos se distribuyen a través de secuencias que dispersan los archivos en los grupos correspondientes (bin). Para evitar que se produzcan condiciones de carrera cuando se escriben varias transmisiones en el mismo archivo, AWK transfiere los nombres de archivo para escribir datos en diferentes lugares, por ejemplo,
chr_10_bin_52_batch_2_aa.csv . Como resultado, se crean muchos archivos pequeños en el disco (para esto utilicé volúmenes EBS de terabytes).
La canalización de la segunda sección
parallel pasa por los grupos (bin) y combina sus archivos individuales en CSV comunes con
cat , y luego los envía para su exportación.
Transmitir a R?
Lo que aprendí : puede acceder a
stdin y
stdout desde un script R y, por lo tanto, usarlo en la tubería.
En la secuencia de comandos Bash, puede observar esta línea:
...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... Traduce todos los archivos de grupo concatenados (bin) en el script R a continuación.
{} es una técnica
parallel especial que inserta cualquier dato enviado por él en la secuencia especificada directamente en el comando mismo. La opción
{#} proporciona una ID de subproceso única y
{%} representa el número de espacio de trabajo (repetido, pero nunca al mismo tiempo). Se puede encontrar una lista de todas las opciones en la
documentación. #!/usr/bin/env Rscript library(readr) library(aws.s3) # Read first command line argument data_destination <- commandArgs(trailingOnly = TRUE)[1] data_cols <- list(SNP_Name = 'c', ...) s3saveRDS( read_csv( file("stdin"), col_names = names(data_cols), col_types = data_cols ), object = data_destination )
Cuando la variable del
file("stdin") se pasa a
readr::read_csv , los datos traducidos en el script R se cargan en el marco, que luego se escribe directamente en S3 como un archivo
aws.s3 usando
aws.s3 .
RDS es un poco como una versión más joven de Parquet, sin los lujos del almacenamiento de columnas.
Después de completar el script Bash, recibí un
.rds archivos
.rds en S3, lo que me permitió usar una compresión eficiente y tipos incorporados.
A pesar de usar el freno R, todo funcionó muy rápido. No es sorprendente que los fragmentos en R que son responsables de leer y escribir datos estén bien optimizados. Después de probar en un cromosoma de tamaño mediano, la tarea se completó en la instancia C5n.4xl en aproximadamente dos horas.
Limitaciones de S3
Lo que aprendí : gracias a la implementación inteligente de rutas, S3 puede procesar muchos archivos.
Me preocupaba si S3 podría manejar muchos archivos transferidos a él. Podría hacer que los nombres de los archivos tengan sentido, pero ¿cómo los buscará S3?
 S3 ,
S3 , / . FAQ- S3., S3 - . (bucket) , — .
Amazon, , «-----» . : get-, . , 20 . bin-. , , (, , ). .
?
: — .
: « ?» ( gzip CSV- 7 ) . , R Parquet ( Arrow) Spark. R, , , .
: , .
, .
EC2 , ( , Spark ). , , AWS- 10 .
R .
S3 , .
library(aws.s3) library(tidyverse) chr_sizes <- get_bucket_df( bucket = '...', prefix = '...', max = Inf ) %>% mutate(Size = as.numeric(Size)) %>% filter(Size != 0) %>% mutate(
, , ,
num_jobs , .
num_jobs <- 7
purrr .
1:1000 %>% map_df(shuffle_job) %>% filter(sd == min(sd)) %>% pull(data) %>% pluck(1)
, . Bash-
for . 10 . , . , .
for DESIRED_CHR in "16" "9" "7" "21" "MT" do
:
sudo shutdown -h now
… ! AWS CLI
user_data Bash- . , .
aws ec2 run-instances ...\ --tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]" \ --user-data file://<<job_script_loc>>
!
: API .
- . , . API .
.rds Parquet-, , . R-.
, ,
get_snp .
pkgdown , .
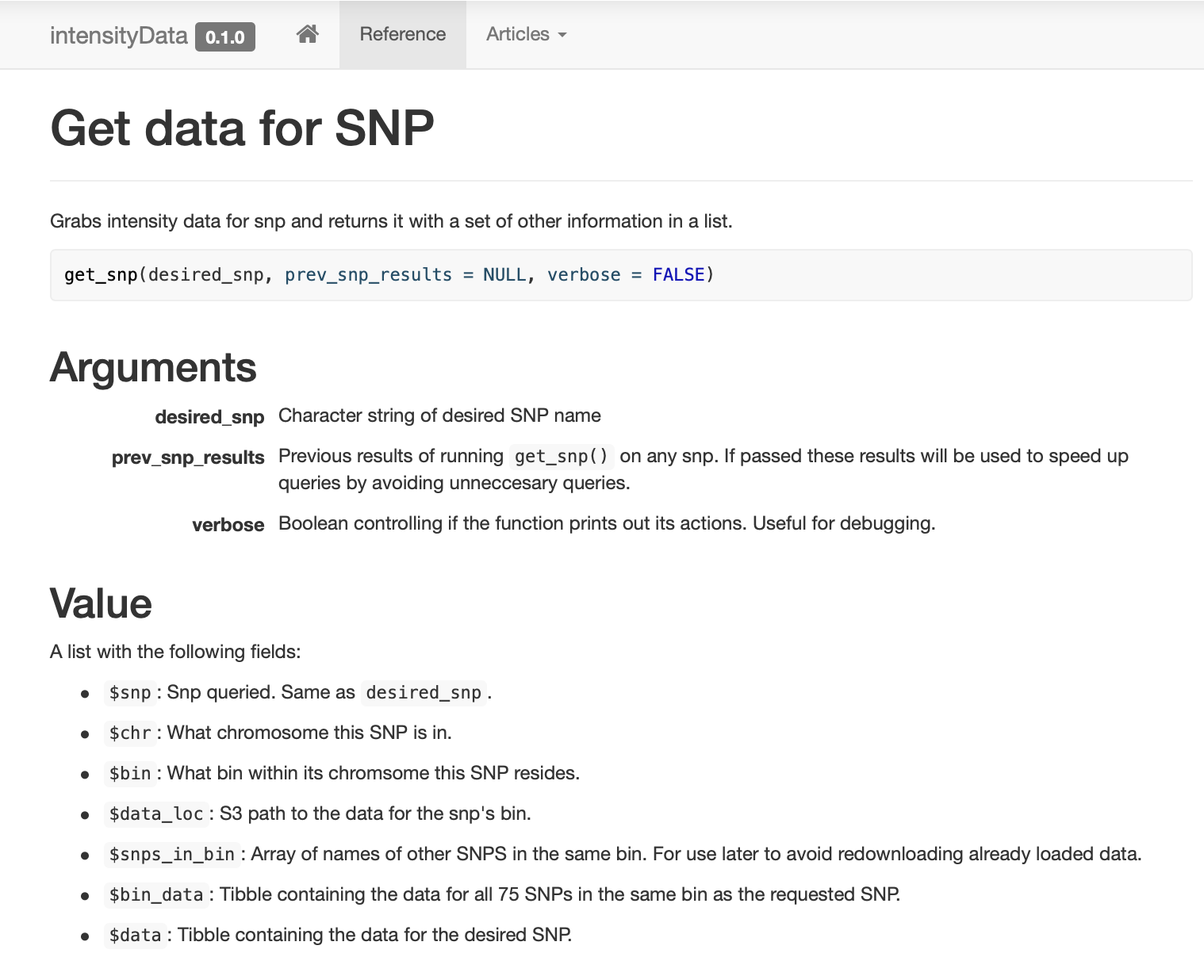
: , !
SNP , (binning) . SNP, (bin). ( ) .
, . , . ,
dplyr::filter , , .
,
prev_snp_results snps_in_bin . SNP (bin), , . SNP (bin) :
Resultados
( ) , . , . .
, , , …
. . ( ), , (bin) , SNP 0,1 , , S3 .
Conclusión
— . , . , . , , , . , , , , . , , , , - .
. , , «» , . .
:
- 25 ;
- Parquet- ;
- Spark ;
- 2,5 ;
- , Spark;
- ;
- Spark , ;
- , , - 1980-;
gnu parallel — , ;
- Spark ;
- Spark ;
- AWK ;
stdin stdout R-, ;
- S3 ;
- — ;
- , ;
- API ;
- , !