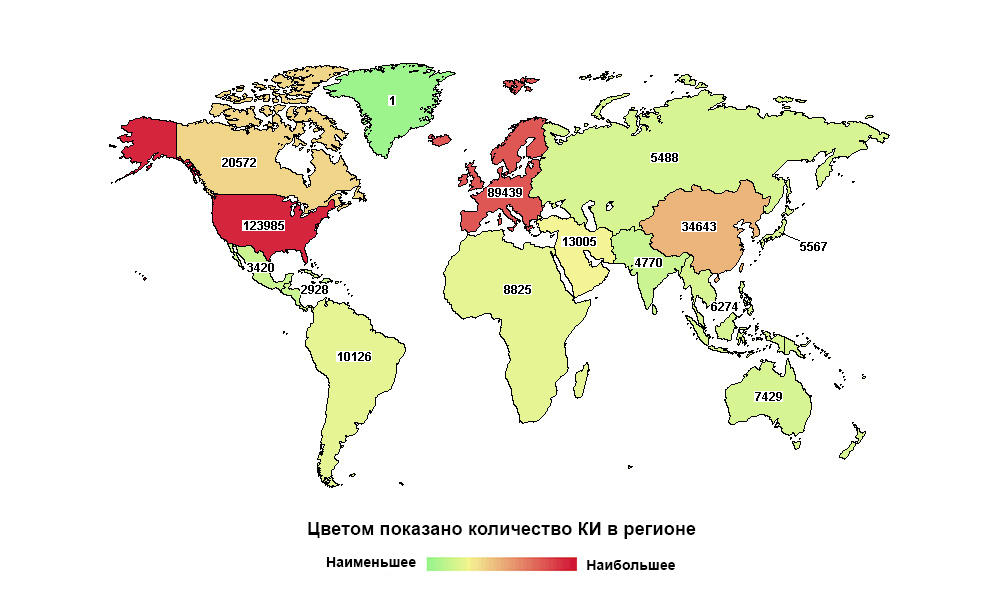
 Rusia está lejos de ser la primera en el mundo, pero la primera en el número de estudios en su macroregión.
Rusia está lejos de ser la primera en el mundo, pero la primera en el número de estudios en su macroregión.Cualquier medicamento hoy, antes de llegar al paciente, pasa por una larga serie de ensayos clínicos. Es necesario demostrar que es capaz de resolver un problema de salud específico y hacerlo de manera más efectiva y, de preferencia, más seguro que sus predecesores.
La selección es estricta: el 98% de todos los medicamentos estudiados no llegan a los pacientes. Con el 2% de los "afortunados", la investigación científica sobre una nueva sustancia antes de ingresar al mercado lleva más de 12 años y más de 1.500 millones de dólares.
Nosotros en la Clínica de
Medicina 24/7 estamos directamente involucrados en ensayos clínicos. Por segundo año consecutivo, llevamos a cabo ensayos clínicos de medicamentos antitumorales extranjeros. Con nuestra ayuda, los nuevos medicamentos tienen acceso a Rusia más rápido y a más de 100 personas al año, otra oportunidad de tratamiento, gratis.
Para las clínicas privadas, la práctica es inusual: un mínimo de beneficios comerciales, demasiadas dificultades para organizar el proceso y requisitos estrictos para una institución médica. Por lo general, solo los grandes centros federales logran cumplir con ellos.
Pero para muchos pacientes en Rusia, un ensayo clínico del medicamento es la única oportunidad de obtener tratamiento gratuito para una enfermedad mortal. Pero entre los pacientes rusos con cáncer, el 30% simplemente no sabe qué es un ensayo clínico, y solo unos pocos participaron en ellos.
Por lo tanto, queremos que la mayor cantidad posible de personas aprendan y comprueben: tal vez tengan la oportunidad de obtener un medicamento que pueda salvarles la vida.
En este artículo explicaremos por qué necesitamos y cómo se organizan los ensayos clínicos, quién y cómo puede llegar allí.
Historias tristes ¿Por qué se necesitan ensayos clínicos y por qué es malo sin ellos?
Investigación / ensayo clínico (en adelante, CI) : un estudio científico que involucra a personas como sujetos, que se realiza para evaluar la efectividad y la seguridad de un nuevo medicamento o ampliar las indicaciones para el uso del ya conocido. Además de los medicamentos, los IC también pueden estudiar la eficacia y la seguridad de los nuevos métodos de tratamiento y diagnóstico.
La medicina está evolucionando y convirtiéndose en una ciencia exacta, que no puede prescindir de las estadísticas.
Anteriormente, el médico de familia conocía las historias de todos sus pacientes de memoria, el médico podía vivir toda su vida en una ciudad, encontrar y recordar un enfoque personal para tratar a todos. Además, la elección de pociones era pequeña: hierbas medicinales, sanguijuelas, mercurio y arsénico. La responsabilidad durante el postulado de "toda la voluntad de Dios" sobre los médicos fue menor.
 Arsénico de finales del XVIII potencia "restaurada" y artritis "curada" ...
Arsénico de finales del XVIII potencia "restaurada" y artritis "curada" ... ... y el mercurio, por ejemplo, era un laxante y "de la sífilis".
... y el mercurio, por ejemplo, era un laxante y "de la sífilis".Cuando la medicina se generalizó, los médicos necesitaban desarrollar tácticas de tratamiento verdaderamente inconfundibles. Se suponía que ciertos medicamentos ayudarían a la mayoría de los pacientes en determinadas condiciones.
Idealmente, un médico debe usar solo aquellos métodos de prevención, diagnóstico y tratamiento que tienen una probabilidad extremadamente baja de obtener "resultados aleatorios", porque su utilidad y efectividad han sido probadas por muchos experimentos realizados correctamente.
Esta es
una medicina basada en evidencia : el único enfoque adecuado para un asunto tan grave como la salud humana en la actualidad.
Y es
la investigación clínica la
base de la medicina basada en la evidencia.
Hasta mediados del siglo XX (!), No había regulación de la investigación sobre nuevos medicamentos. Para restablecer el orden, como suele suceder, se necesitaron un par de tragedias.
En 1937, 105 niños y un adulto murieron, tomando el "elixir" de la sulfonamida antiséptica y ... dietilenglicol venenoso. Sí, el que se usa hoy en anticongelante. Luego, la compañía farmacéutica
ME Massengill, sin saberlo, lo usó como solvente, excipiente. No se han realizado estudios de seguridad del "cóctel" resultante para humanos. Cuando de repente se dieron cuenta y tomaron la droga de la venta, ya había más de cien víctimas. En 1938, el Congreso de los Estados Unidos aprobó una ley sobre la investigación obligatoria de drogas antes de que salgan a la venta. El control sobre esto fue confiado a la FDA (
Administración de Drogas y Alimentos en inglés), la Administración de Drogas y AlimentosUn escándalo aún más fuerte ocurrió con la
talidomida a fines de los años cincuenta y principios de los sesenta. "Las pastillas calmantes y para dormir, que ayudan perfectamente con la toxicosis de las mujeres embarazadas" se agotaron rápidamente. Sus estudios se llevaron a cabo solo en ratas. Resultó que esto no es suficiente. En humanos, la talidomida causó defectos en el desarrollo del feto. En Europa, Australia y Japón, alrededor de 10,000 niños nacieron con malformaciones (malformaciones) de las extremidades. La droga fue prohibida en la mayoría de los países en 1961.
 Las madres de estos niños tomaron pastillas para dormir, no probadas en humanos
Las madres de estos niños tomaron pastillas para dormir, no probadas en humanosDesde entonces, los medicamentos han sido cuidadosamente estudiados antes del registro. Esto se rige por las
Reglas tripartitas armonizadas internacionales de buenas prácticas clínicas (Directriz tripartita armonizada ICH para buenas prácticas clínicas, abreviado como ICH GCP). Desde 1996 hasta 1997, operan en los Estados Unidos, Japón y la UE, y desde 2003 se han introducido en Rusia.¿Cómo va el estudio y por qué durante tanto tiempo?
Todo el proceso de creación del medicamento se puede dividir en 3 grandes fases.
1. Búsqueda de ideas y estudios preclínicos, in vitro y en animales.
2. Si esto no terminó allí, entonces comienzan los estudios clínicos, con personas: primero cuidadosas, luego más masivas.
3. El medicamento se registra con las autoridades reguladoras para convertirse en un nombre familiar en los directorios médicos.
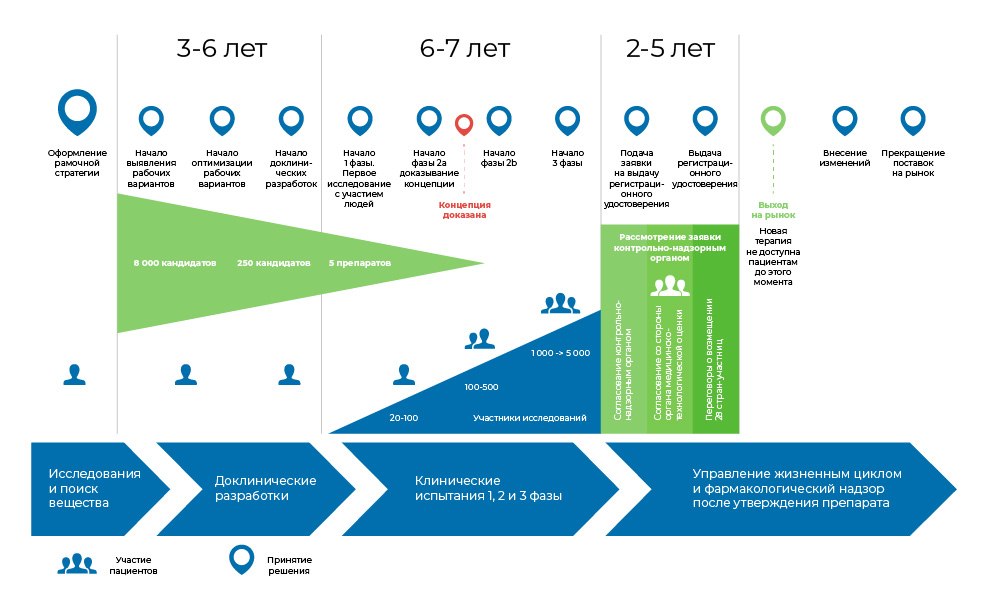 El proceso de desarrollo de una droga. Desde el momento en que se crea la molécula hasta el inicio de la venta del medicamento, se requieren de 8 a 20 años.Entonces, ¿alguien necesita esto?
El proceso de desarrollo de una droga. Desde el momento en que se crea la molécula hasta el inicio de la venta del medicamento, se requieren de 8 a 20 años.Entonces, ¿alguien necesita esto? La oncología es una de las áreas más notorias de la medicina en términos de la necesidad insatisfecha de medicamentos. Según la Organización Mundial de la Salud, en 2018, el cáncer
mató a 9,6 millones de personas. Los tumores a menudo se encuentran en las etapas posteriores, cuando solo queda el tratamiento paliativo.
Al mismo tiempo, los descubrimientos en el campo de la biología molecular y la genética permitieron comprender los mecanismos que contribuyen al desarrollo y la progresión del cáncer, y mejoraron la comprensión del trabajo de la inmunidad antitumoral.
Y hoy, el desarrollo de medicamentos antitumorales es una de las áreas de medicina más intensivas en ciencia y más populares.
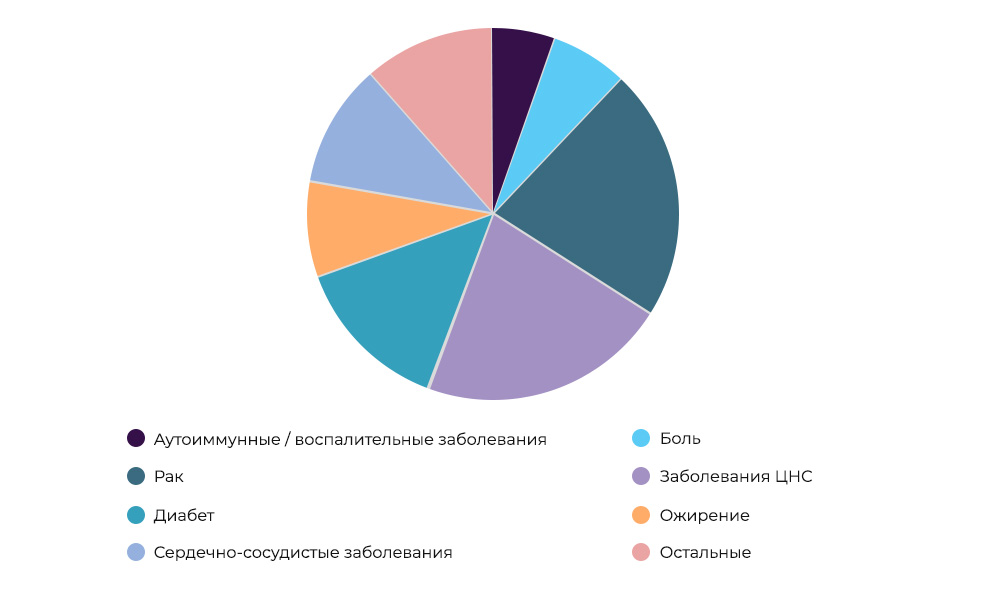 Investigación sobre medicamentos contra el cáncer: el 23% de todas las IC en el mundoQuién paga por la investigación.
Investigación sobre medicamentos contra el cáncer: el 23% de todas las IC en el mundoQuién paga por la investigación. A veces, el organizador y el patrocinador pueden ser una organización de investigación. Pero con mayor frecuencia, los científicos se dedican a la investigación científica a expensas de las compañías farmacéuticas. Aquellos esperan ingresar al mercado con un medicamento exitoso, obtener ganancias y recuperar los costos de CI y desarrollo. Es como comprar una nueva película para alquilar en una película: el distribuidor no sabe si "filmará" o no. Producir nuevos medicamentos es un negocio muy arriesgado.
Anteriormente, muchas compañías farmacéuticas realizaban investigaciones por su cuenta, utilizando el personal de los científicos. Ahora, una institución médica que ha aprobado la acreditación y cumple con ciertos requisitos puede convertirse en una plataforma e intérprete para el experimento.
Este es exactamente el caso de nuestra
"Medicina 24/7" . La compañía farmacéutica está lista para pagar, pero las finanzas se asignan al final del estudio, en función de los costos incurridos. La clínica no gana ninguna superganancia. Y los médicos de investigación generalmente no ganan nada además de su salario habitual. Más bien, esta es la posición del jefe de la clínica: una persona considera que es correcto hacer avanzar la medicina del país y aprovecha la oportunidad para participar en esto.
Primero surge una idea. En realidad, ¿qué investigar? En oncología, primero se encuentran "objetivos": un punto débil de la enfermedad. Si interrumpe o simplemente "apaga" las moléculas objetivo, el tumor "sufre". Muchos medicamentos modernos contra el cáncer,
dirigidos o
inmunoterapéuticos, se basan en este principio.
 El mecanismo de las drogas dirigidas en el cáncer colorrectal. Las células cancerosas dejan de dividirse, o hacen crecer vasos sanguíneos adicionales al tumor, o el medicamento protege a las células vecinas de convertirse en malignas
El mecanismo de las drogas dirigidas en el cáncer colorrectal. Las células cancerosas dejan de dividirse, o hacen crecer vasos sanguíneos adicionales al tumor, o el medicamento protege a las células vecinas de convertirse en malignasPara encontrar tales sustancias, y luego elegir los candidatos más adecuados, se requieren muchos recursos y tiempo para
estudios in vitro y en silicio , es decir, in vitro o mediante simulación por computadora.
La sustancia seleccionada se almacena en la cantidad correcta, producida de acuerdo con reglas especiales (en Rusia es GOST R 52249-2009), sin impurezas y violación de la tecnología. Y con estos tubos de ensayo, los científicos van a probar la droga en animales.
El mouse es el motor del progreso. Después de probar ideas in vitro, un científico con un suministro de su medicamento potencial va al vivero: debe verificar cómo se comporta el prototipo en el cuerpo de un mamífero (in vivo).
Incluso en 1025, Avicena escribió en el "Canon de la ciencia médica" que los medicamentos deben ser controlados. Además, es deseable, en un paciente potencial, una persona. Después de todo, el resultado obtenido en leones y caballos no garantiza que el medicamento afecte a las personas de la misma manera.
Y aún en medicina, sin experimentos con animales, no se puede hacer. Los leones y los caballos, sin embargo, se quedaron solos. Los estudios preclínicos en todo el mundo ocurren principalmente en ratones, cobayas y conejos.
 Los ratones de laboratorio incluso pusieron un monumento en el Novosibirsk Academgorodok
Los ratones de laboratorio incluso pusieron un monumento en el Novosibirsk AcademgorodokEn esta etapa, verifique qué tan dañino / seguro es el medicamento:
- ¿Causa alergias?
- ¿Tiene efectos tóxicos en los tejidos y órganos,
- cómo afecta la capacidad de los animales para reproducirse y el desarrollo normal del feto, etc.
Además, observan cómo se comporta el candidato a fármaco dentro del cuerpo del animal (farmacocinética):
- tasa de absorción y aumento de la concentración sanguínea,
- ¿Cuáles son las dosis máxima y mínima?
- qué tan rápido se excreta del cuerpo, etc.
Todos estos datos son necesarios para decidir
si es posible usar la sustancia de prueba para humanos. Y si es así, cuánto se necesita.
El mal inevitable. Burocracia El
Departamento de Estado supervisa el progreso correcto de CI
. regulación de la circulación de medicamentos del Ministerio de Salud y el Servicio Federal de Supervisión de la Atención Médica (Roszdravnadzor).Si el científico ha llegado al momento en que es necesario proceder a los ensayos clínicos en humanos, es hora de preparar una solicitud para realizar IC. Para hacer esto, necesita varios
documentos .
- Dossier del fármaco del estudio. Todo lo que ya se ha descubierto sobre el medicamento: datos sobre farmacocinética, efectividad, toxicidad, etc.
- Protocolo de estudio Detalla el plan para futuras investigaciones y métodos para evaluar los resultados;
- Folleto del investigador. Una breve hoja de trucos para explicar claramente la esencia del estudio a voluntarios y pacientes y obtener su consentimiento informado.
Comité de ética. La siguiente etapa de la búsqueda es obtener la evaluación y la conclusión del comité de ética.
El Comité de Ética es un grupo independiente de médicos, científicos, personal médico y no especialistas (miembros del público). Estudian el protocolo de investigación y el consentimiento informado para asegurarse de que haya un acuerdo entre el paciente, los investigadores, la compañía farmacéutica y la autoridad reguladora nacional, que no se violen los derechos de nadie, que nadie esté sujeto a coerción y que nadie haya infringido el libre albedrío.
Después de los efectos secundarios graves de un medicamento en 2006, el comité de ética se volvió aún más estricto. Por lo tanto, a veces el estudio puede "congelarse" en esta etapa durante un año o más.
Después de verificar todos los documentos y la aprobación del comité de ética, el medicamento potencial pasa a la etapa de
ensayos clínicos, en humanos.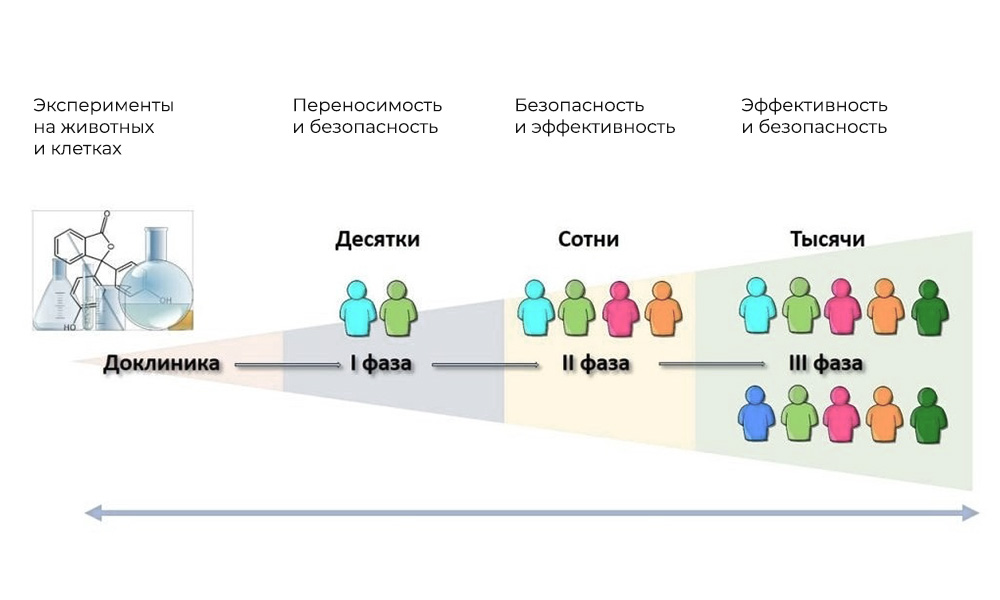 Las principales fases de los ensayos clínicos son en humanos.
Las principales fases de los ensayos clínicos son en humanos.Fase I. Prueba del mecanismo de acción.
Participantes: 20 - 100 personas.
Duración: de varios meses a 1 año.
Objetivo: estudiar tolerancia, farmacodinámica y farmacocinética.
Se verifica si la sustancia actúa en humanos de la misma manera que en animales, si es segura.
En la primera fase de un estudio clínico, en teoría, los voluntarios sanos deberían participar, pero en las pruebas oncológicas de sustancias potentes en un cuerpo sano no se puede llamar ético. Por lo tanto, las personas con la enfermedad correspondiente están involucradas, contra las cuales el futuro medicamento puede ser efectivo.
A los participantes se les inyectan gradualmente todas las dosis más grandes del medicamento, comenzando desde el mínimo hasta el máximo permitido. Después de cada administración, se monitorea la condición del paciente.
Se evalúa la farmacocinética : tasa de absorción y excreción (excreción de la sustancia inalterada), distribución sobre tejidos y órganos.
La farmacodinámica también se evalúa: el efecto del medicamento sobre las células tumorales, sobre otros órganos y órganos, los efectos secundarios. La aplicación preferida y el nivel de dosificación se están aclarando.
Además de los estudios con dosis crecientes, en la fase I verifico:
- el efecto de la comida en la droga;
- interacción con otras drogas;
- El efecto de otras enfermedades que pueden afectar la dosis deseada del medicamento (por ejemplo, en un paciente con insuficiencia renal).
Según la
FDA , el 70% de los medicamentos pasan con éxito la primera fase de CI.
Fase II Verificación de la acción para un objetivo dado: un tipo específico de enfermedad
Participantes: de 100 a 500 pacientes.
Duración: de varios meses a 2 años.
Propósito: probar la efectividad de ciertas indicaciones
Es necesario estudiar qué tan efectivo es el nuevo medicamento en comparación con el placebo o el tratamiento existente. Además, un mayor número de participantes puede detectar efectos secundarios más raros que no se detectan en la fase I.
Para participar en esta fase de IC, los pacientes se seleccionan de acuerdo con un número de criterios mucho mayor que en la primera fase. Por ejemplo, no solo “cáncer de seno”, sino “cáncer de seno, etapa T2N1M0, subtipo HER2 positivo”.
Por lo general, los estudios en esta etapa se realizan como
doble ciego, aleatorio, controlado con placebo.Doble cegamiento: ni el médico ni el paciente saben quién está recibiendo el principio activo y quién está recibiendo un placebo o el tratamiento óptimo que existe actualmente.
La aleatorización implica que los pacientes se dividen en grupos al azar, utilizando un generador de números aleatorios. Ni el médico ni el participante en CI pueden influir en este proceso.
El control de placebo significa que los participantes en un grupo recibirán un placebo en las mismas condiciones que los participantes en otro grupo que reciben la sustancia activa.
 Todos: la misma apariencia, sabor y olor de la medicina.
Todos: la misma apariencia, sabor y olor de la medicina.Toda esta "teología de la conspiración" es necesaria para excluir la distorsión intencional o inconsciente de los datos experimentales por parte de los participantes o investigadores.
Entonces, en la primera fase, donde no hay requisitos tan estrictos, hay resultados sorprendentes. Esta es una estadística "sucia": en la fase II se elimina el exceso y los resultados se vuelven plausibles.
Según la FDA, solo el 33% de los medicamentos que alcanzan la fase II se someten con éxito a CI y pasan a la siguiente fase.
Fase III Estudios de apoyo
Número de participantes: 300 - 3.000 o más.
Duración: de un año a varios años.
Propósito: confirmación de la efectividad y seguridad de la sustancia de prueba en muestras grandes.
Esta es la parte más grande, más compleja y más costosa del proceso de desarrollo de fármacos. El propósito de tales estudios es confirmar la efectividad y seguridad de la sustancia de prueba cuando es utilizada por un gran número de pacientes.
Según los resultados de esta fase, los fabricantes de medicamentos reciben permiso para llevarlo al mercado.
En la fase III, pueden participar miles de pacientes de diferentes países. Todo debe planificarse hasta el más mínimo detalle, para que en todos los lugares del estudio su diseño y condiciones significativas sean exactamente las mismas.
El diseño del estudio es tan estrecho que no solo un paciente moribundo, sino también un paciente con un pronóstico de remisión estable puede entrar en él. El medicamento debe ser tan seguro que se pueda administrar a una persona prácticamente sana, y la calidad de vida no disminuye.
Antes del comienzo de la fase III, hay muchas consultas y discusiones entre investigadores y expertos externos: es muy importante pensar en el diseño de experimentos para no perder lo importante y obtener todos los datos necesarios.
Durante la fase III, la eficacia y seguridad del nuevo fármaco y la relación dosis-respuesta finalmente se confirman.
Se analiza la correlación de ventajas y riesgos. En función de los resultados, el organismo regulador decide si es posible llevar el medicamento al mercado. Para hacer esto, se deben cumplir las siguientes condiciones:
- la droga es más efectiva que los análogos conocidos previamente,
- da menos efectos secundarios / mejor tolerado
- eficaz cuando las drogas previamente conocidas no funcionan,
- más rentable económicamente,
- Más fácil de usar.
El proceso de revisión de la solicitud por parte del supervisor demora entre 12 y 18 meses.
Según la FDA, la tercera fase de los ensayos clínicos termina con un resultado positivo en solo el 25-30% de los casos de todos los que estaban al comienzo de la tercera fase.
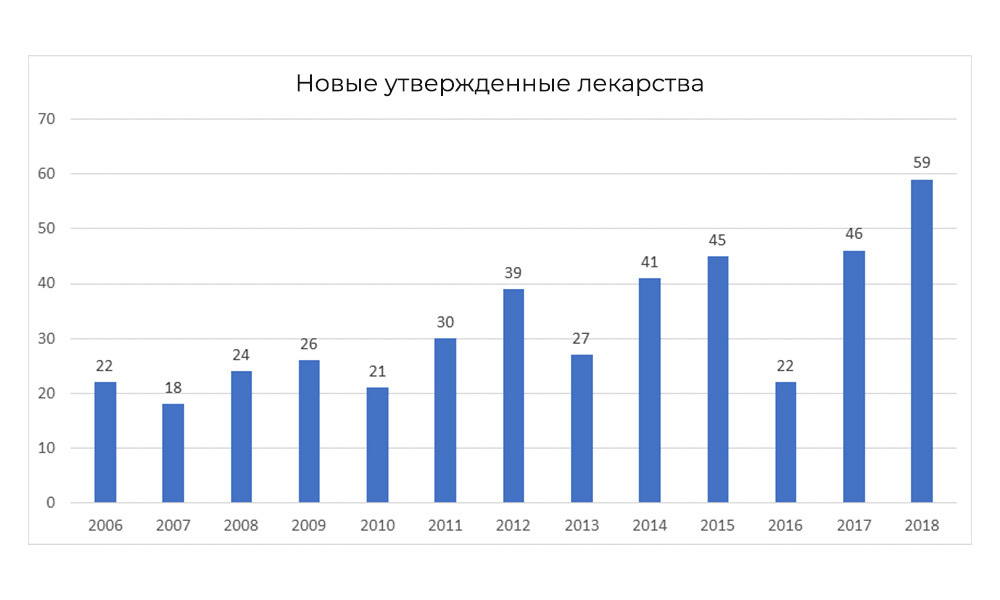 Sin embargo, en 2018, la FDA rompió su propio récord de cantidad de medicamentos aprobados.
Sin embargo, en 2018, la FDA rompió su propio récord de cantidad de medicamentos aprobados.Características de la investigación nacional: una fase adicional de CI en Rusia
El control de nuevas drogas en Rusia tiene sus propios errores (o características, cómo verse). Según la ley, los medicamentos extranjeros aprobados deben someterse a ensayos clínicos adicionales en Rusia: supuestamente, esto aumentará la calidad de los medicamentos extranjeros.
3 , , . , , , 3 , , .
, 12 – 12 , . , 6 12. , , , , .
2-3 .
« 24/7» III.
. , « ». , . , – , , .
, III – – , «» .
, . .
-,
GCP, Good Clinical Practice.-, . , - : - . . .
-, . , , , , , , – . – : , .
 –
–, , , , .
1 200 , . : , , , , . « » – - .
2 3 , .
2 :
. , , , , .. —
. .
– , , .
, . «», : , , / , – .
. , – . , .
18 .
, – , , 3-4 . : 10 , – .
, , .
– – . , . , , .
, , , , – – . .
, .
–
RosOncoWeb ,
CTAgency .
« 24/7» – .