En el
artículo detallado anterior sobre el
Genoma Completo, prometimos publicar tres problemas y evaluar al primero que resuelva los tres correctamente. Al mismo tiempo, damos ejemplos de cómo trabajar con datos genéticos en estas tareas. Hoy publicamos el primero.

En el primer
artículo, compartimos información útil y enlaces que son útiles para trabajar con datos bioinformáticos. Le recomendamos que lo lea primero si se lo perdió.
Descargo de responsabilidadEl trabajo con datos genéticos se lleva a cabo en sistemas Unix (Linux, macOS), ya que algunos comandos y software no están disponibles en Windows. Por lo tanto, para los usuarios de Windows, una de las soluciones más simples es alquilar una máquina virtual Linux.
Todas las operaciones descritas a continuación se realizan en la línea de comando - terminal. Antes de comenzar, aprenda a trabajar en una terminal que ejecute su sistema operativo y use comandos, ya que algunos de ellos pueden dañar el sistema operativo y sus datos.
Software requerido
Hemos recopilado la
imagen de una máquina virtual (VM) con todo el software necesario en Yandex.Cloud. Regístrese en Yandex.Cloud, en su cuenta en la sección Compute Cloud, haga clic en Crear VM. Como imagen pública, seleccione 1000 Genomas del catálogo de Atlas Data Analysis.
Configuración de VM: 100% 2vCPU, 8GB RAM, 20GB HDD. Al crear una VM, debe ingresar los datos de pago, pero no se elimina nada de la cuenta. Un inicio y una subvención adicional en una palabra de código es suficiente para trabajar con una VM e imagen de Atlas hasta el 31 de diciembre de 2019 de forma gratuita. Para recibir una subvención para completar tareas, envíe la palabra de código "ATLAS" al
soporte de Yandex.Cloud .
Nota: la subvención es válida para los nuevos usuarios de Yandex.Cloud que se hayan registrado desde el 18 de diciembre de 2019 o para aquellos que todavía tienen un período de prueba y tienen una subvención inicial. La palabra de código ATLAS es válida solo una vez.
Primero cree una clave ssh en la computadora local desde la que planea conectarse a la VM:
ssh-keygen -o -t rsa -b 4096 -C "my-local-machine" -f ~/.ssh/yandex-cloud -a 100
No olvide copiar el contenido del archivo
~/.ssh/yandex-cloud.pub en la ventana correspondiente al crear la VM.
Si desea instalar el software en su computadora, a continuación encontrará toda la información de instalación. Si decide usar Yandex.Cloud, cree una VM y continúe con la siguiente sección.
Plink
Plink es un paquete de software para manipular datos genéticos y la búsqueda de asociación de genoma amplio (GWAS). Fue desarrollado por el genetista Sean Purcell (Shaun Purcell). Desde 2008, con la ayuda de Plink, se han realizado cientos de GWAS en todo el mundo, cuyos resultados son los mejores que Atlas utiliza como fuente de datos para algoritmos para calcular riesgos de enfermedades.
Plink ofrece un conjunto de herramientas para almacenar y convertir datos de genotipado y buscarlos. Plink también permite el procesamiento estadístico, análisis de desequilibrio de ligamiento (LD), análisis de identidad por descendencia (IBD) e identidad (estado por IBS), estratificación de la población y pruebas de epistasis: la interacción de varias variaciones genéticas entre ellos
La EII y el SII se utilizan para analizar la composición de la población y determinar el parentesco.
Un ejemplo de epistasis es la variación de rs7412 y rs429358 en el gen APOE, una cierta combinación de variantes que aumenta drásticamente el riesgo de desarrollar la enfermedad de Alzheimer, mientras que cada variante individualmente solo hace una pequeña contribución al riesgo.
Descargue la versión estable de Plink desde el sitio
web oficial.
BCFtools
BCFtools es un conjunto de utilidades para manipular datos genéticos en el formato VCF y su contraparte binaria BCF. La lista de posibles aplicaciones de BCFtools incluye anotación, filtrado, fusión y división de archivos VCF / BCF, encontrar sus intersecciones, indexación, búsqueda selectiva, clasificación, conteo de estadísticas, etc.
Para instalar, haga:
git clone git://github.com/samtools/htslib.git git clone git://github.com/samtools/bcftools.git cd bcftools
El proceso de instalación se describe con más detalle
aquí .
REY
El paquete KING (inferencia basada en el parentesco para Gwas) se utiliza en estudios de población cuando se trabaja con datos de una búsqueda de asociaciones en todo el genoma para determinar las relaciones de parentesco en los datos estudiados. En esta tarea, KING ayudará a determinar el grado de parentesco de varias muestras del proyecto 1000 Genomes.
Puedes descargarlo
aquí . Para resolver problemas, el manual de KING está disponible
aquí .
Casi todos los errores que pueden surgir durante el trabajo con herramientas se describen en Stackoverflow o su contraparte bioinformática: Biostars .
Datos utilizados
Para orientación, utilizamos los datos abiertos
del proyecto 1000 Genomes. Para el análisis, seleccionamos 10 muestras con información sobre los genotipos de aproximadamente 85 millones de variaciones obtenidas mediante el análisis de datos NGS alineados con la versión del genoma de referencia GRCh37. Las relaciones familiares y las poblaciones de muestra se muestran en la Figura 1.
 Figura 1
Figura 1 Pedigree utilizado en muestras de VCF. El cuadrado corresponde al género masculino, el círculo al femenino. Línea punteada significa parentesco de segundo orden indeterminado.
Toma nota
El formato VCF le permite almacenar información sobre el campo de una persona como un solo número, si esta información se conocía durante la generación de VCF. Se ve así: el campo GT (genotipo, genotipo) para registros del cromosoma X contiene un valor numérico que corresponde a un alelo, para hombres y dos para mujeres. Si no hay información sobre el campo biológico de la muestra secuenciada, entonces el campo GT contendrá por defecto dos valores numéricos (resaltados en rojo en la Figura 2).
En los archivos VCF utilizados en este manual, se excluye el cromosoma Y, pero la presencia del cromosoma Y en el archivo VCF no siempre significa que la muestra secuenciada realmente lo tenga. Esto se debe a las regiones pseudo-autosómicas (PAR), que son idénticas para los cromosomas X e Y y se encuentran en sus extremos.
Los cromosomas diferentes normalmente no tienen regiones idénticas largas (homólogas), sin embargo, los cromosomas X e Y tienen tales regiones de varios millones de pares de bases de largo al principio (PAR1) y al final (PAR2). Por lo tanto, al analizar los datos de NGS en hombres en las regiones PAR, se encuentran dos alelos (uno para cada cromosoma sexual), y en las mujeres, los genotipos pueden aparecer en las regiones PAR del cromosoma Y, aunque en realidad son genotipos de su cromosoma X.
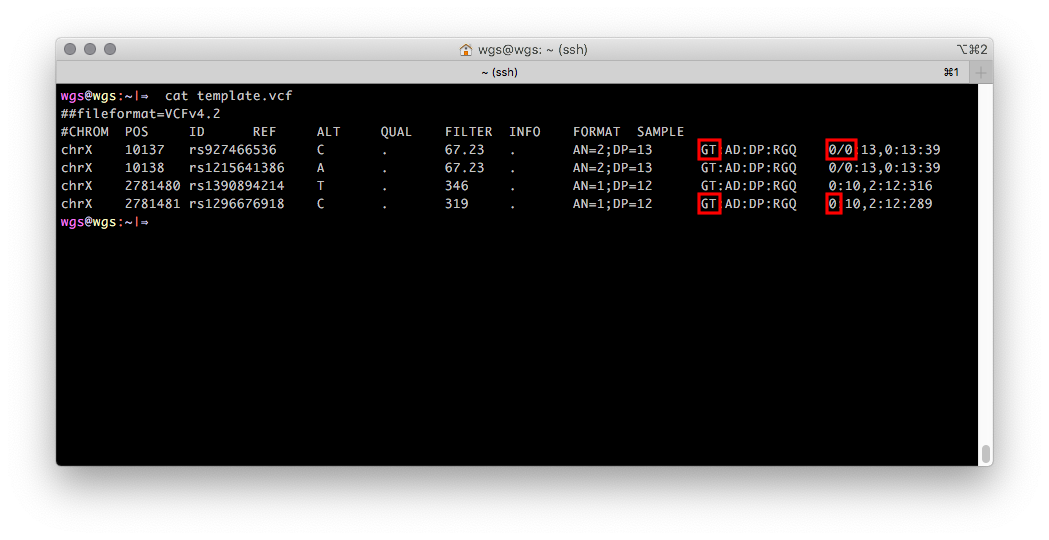 Figura 2
Figura 2 Archivo VCF con genotipos del cromosoma X de un hombre de la región PAR1 (dos primeras entradas) y región no pseudo-autosómica (dos últimas entradas).
Unidad educativa
El género genético es un conjunto de cromosomas sexuales que corresponden a la manifestación de características sexuales primarias y secundarias de un tipo masculino o femenino. Normalmente, los hombres tienen un cromosoma X y un cromosoma Y, mientras que las mujeres tienen dos cromosomas X. Con diversos trastornos en la formación de células germinales, óvulos y espermatozoides, un niño con un excelente conjunto de cromosomas sexuales puede nacer de los padres, lo que a menudo conduce a trastornos del desarrollo. características sexuales primarias y secundarias.
Las dos anomalías sexuales cromosómicas más comunes son el síndrome de Turner (un conjunto de cromosomas X0, es decir, solo un cromosoma X) y el síndrome de Klinefelter (un conjunto de cromosomas XXY).
Un alelo es uno o más nucleótidos que se encuentra en cualquier posición del genoma y tiene una alternativa. El concepto se usa para describir genotipos. Distinguir entre alelos de referencia y alternativos. Todos ellos se almacenan en el archivo VCF en los campos REF y ALT, respectivamente.
Determinar el género
Para usuarios de Yandex.CloudTodos los datos para completar el manual y las tareas independientes se almacenan en Yandex.Cloud utilizando la estructura que se muestra a continuación. La carpeta
Tutorial contiene el archivo VCF necesario para completar el manual, la carpeta
Test para tareas independientes. La carpeta
Technical contiene dos archivos con una lista de identificadores de variaciones genéticas:
rsids_for_subsetting.txt usa en el manual y las tareas para la ejecución independiente, es posible que se necesite
external_interpretation_rsids.txt en el futuro cuando se adquiere la secuenciación del genoma completo en el Atlas para cargar datos de genotipado a servicios de terceros. La carpeta
Tools contiene, entre otras cosas, dos scripts utilizados en las tareas 2 y 3.
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
Se creará una carpeta en el directorio
/home en la máquina virtual Yandex.Cloud, cuyo nombre corresponde al nombre de usuario especificado en la etapa de creación de la máquina virtual. Copie todo desde el directorio
/home/ubuntu a su directorio a través de los siguientes comandos:
cd ~ cp -r /home/ubuntu/* ./
Por lo demásCuando trabaje en una PC personal, puede descargar los archivos necesarios para la primera tarea desde el
enlace . El archivo descargado admite una estructura de almacenamiento de archivos similar a la utilizada en Yandex.Cloud:
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
Descomprima el archivo
atlas_wgs_contest.tar.gz con el comando
tar -xvzf atlas_wgs_contest.tar.gz Los archivos VCF para realizar tareas en forma no archivada ocupan aproximadamente 19 gigabytes cada uno, por lo tanto, para ahorrar espacio, recomendamos trabajar solo con archivos. Todos los programas enumerados anteriormente ya pueden trabajar con datos VCF comprimidos. Además, no necesita hacer nada.
Para determinar el sexo del sujeto, debe observar los genotipos en el cromosoma X y excluir las regiones PAR1 y PAR2 ubicadas al principio y al final. Estos son los intervalos de las posiciones 60001–2699520 y 154931044–155260560 en la versión GRCh37 del genoma. Si el genotipo contiene una designación numérica, este es el sexo biológico masculino, de lo contrario, el femenino. Debe tenerse en cuenta que la designación de género en el archivo VCF depende de la disponibilidad de información sobre el campo biológico durante la generación de VCF, por lo tanto, este enfoque no siempre se puede utilizar.
Use el siguiente comando para cada una de las muestras en el conjunto de datos. Sustituya el identificador de muestra después del argumento
-s :
(/Data/Tutotrial/CEI.1kg.2019.demo.vcf.gz):
Al ejecutar los comandos, verá una parte del contenido del archivo VCF para el identificador de muestra especificado. El
-r chrX:2699521-154931043 en BCFtools restringe la visualización del contenido del archivo a la región del cromosoma X desde la posición 2699521 a la posición 154931043 (región no PAR en la Figura 3). Estos límites excluyen regiones pseudo-autosómicas innecesarias en este caso (PAR1 y PAR2). Usando los valores numéricos en el campo GT, determine el género de cada muestra.
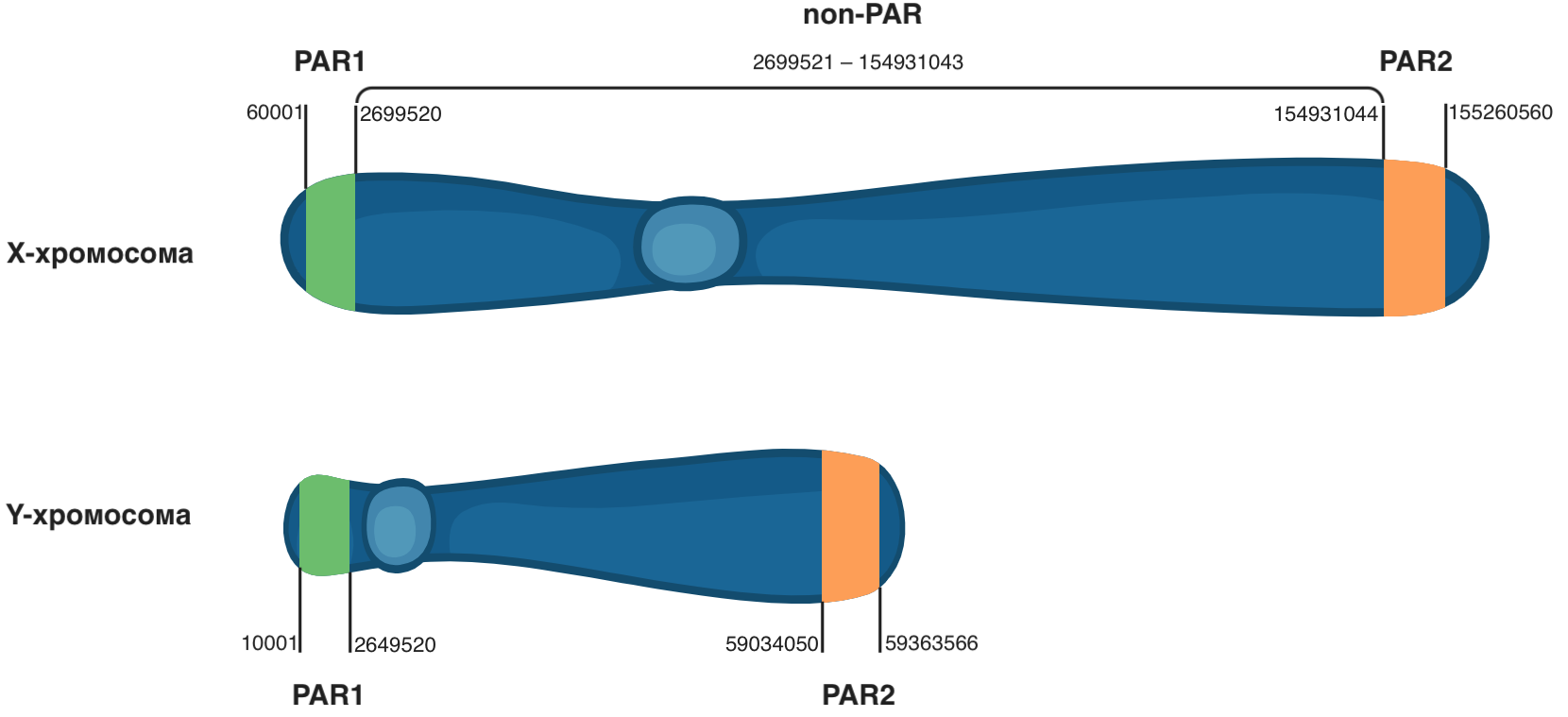 Figura 3
Figura 3 La ubicación de las regiones pseudo-autosómicas de PAR1 y PAR2 en los cromosomas sexuales.
Puede ver la lista de todos los identificadores de muestra en el archivo VCF en la Figura 1 o en la última línea del encabezado del archivo VCF. Se enumerarán después del nombre de la columna FORMATO:
El verdadero género de estas muestras también se muestra en la Figura 1.
Determinamos la relación
Para determinar la relación, necesitamos comparar por pares los datos genéticos de todas las muestras. Es difícil hacer esto de acuerdo con el genoma completo: en este caso, un archivo VCF toma decenas de gigabytes. El VCF que utilizamos ocupa solo alrededor de 2 gigabytes, pero aún lo filtramos de acuerdo con la lista de identificadores de variación genética (rsID) genotipados en chips de Illumina: GSA v1, GSA v2, HumanOmniExpress v1.0, HumanOmniExpress v1.3, InfiniumExome v1. 1 e Infinium OmniExpressExome v1.4. Estos son los chips más populares en genotipado comercial.
Hemos compilado una lista de todos los identificadores de variaciones genéticas de estos chips en un archivo separado con una lista de rsID. Contiene 1,4 millones de identificadores. Para filtrar el archivo VCF, ejecute el siguiente comando:
bcftools view -O z -i 'ID=@rsids_for_subsetting.txt' CEI.1kg.2019.demo.vcf.gz > CEI.1kg.2019.demo.subset.vcf.gz
Cada vez que utiliza BCFtools y otros paquetes para trabajar con archivos VCF, el historial de comandos anteriores se agrega al encabezado del archivo. Independientemente del método de filtrado del archivo VCF y los comandos ejecutados anteriormente, puede verificar la integridad e identidad de los contenidos principales del VCF calculando la suma de hash:
El comando
gunzip -c descomprime el archivo y
gunzip -c su contenido en stdout, desde donde se eliminan las líneas de encabezado del archivo VCF que comienzan
# (por lo tanto, se utiliza el
grep -v "^#" ). El encabezado se elimina para comparar la integridad de solo los datos genéticos en sí, y no los metadatos sobre qué herramientas y cuándo se usaron para trabajar con este archivo VCF.
Si el valor hash coincide, puede continuar y convertir VCF al formato Plink interno (de forma predeterminada, el formato Plink es tres archivos con las extensiones bed, bim y fam). En estos archivos, solo queda el genotipo, el cromosoma, la posición y algunos otros datos, y el resto se elimina. Con este formato es mucho más fácil trabajar y resolver varios problemas que no requieren información adicional de VCF. Por ejemplo, realizar GWAS.
Este comando creará tres archivos en la carpeta:
CEI.1kg.2019.demo.subset.bed
CEI.1kg.2019.demo.subset.bim
CEI.1kg.2019.demo.subset.famPuede determinar el parentesco en parejas para las 10 muestras. Utilizamos el siguiente comando para analizar archivos Plink:
king -b CEI.1kg.2019.demo.subset.bed --kinship --prefix CEI.1kg.2019.demo.subset.kinship_analysis
Mire el archivo
CEI.1kg.2019.demo.subset.kinship_analysis.kin0 y preste atención a la columna Kinship, que contiene los coeficientes de parentesco para los pares de muestras indicados en ID1 e ID2, respectivamente.
Compare los coeficientes que obtuvo en el archivo
CEI.1kg.2019.demo.subset.kinship_analysis.kin0 para todos los pares de muestras con el pedigrí que se muestra en la Figura 1 (la línea discontinua corresponde al parentesco de segundo orden, sin embargo, no hay datos exactos de parentesco, es decir, esto puede haber primos, tía / sobrino o tío / sobrina). Trate de llegar a su propia conclusión sobre qué valores de los coeficientes de parentesco pueden corresponder a la relación de primer y segundo orden.
PistaExtracto de la documentación de KING: los coeficientes de parentesco> 0.354 corresponden a muestras duplicadas o gemelos idénticos, de 0.177 a 0.354 a parentesco de primer orden (padres-hijos, hermanos), de 0.0884 a 0.177 a parentesco de segundo orden (primos, tías / tíos-sobrinos), y de 0.0442 a 0.0884 - al parentesco de tercer orden (abuelos, nietos, primos segundos). Cualquier cosa menor que 0.0442 es difícil de interpretar sin ambigüedades.
La primera tarea de la competencia.
Utilizando un conjunto de datos de prueba de 12 muestras
Data/Test/CEI.1kg.2019.test.vcf.gz ,
Data/Test/CEI.1kg.2019.test.vcf.gz su pedigrí, guiado por los resultados de la determinación del sexo y el análisis de parentesco. Las muestras que, según los resultados del análisis, no tienen parentesco con alguien, anote cerca, sin conectarlas con una línea con otras muestras. El pedigrí puede estar compuesto en un estilo similar a la Figura 1, sin embargo, esto queda a su discreción. Los hombres están indicados por un cuadrado, las mujeres por un círculo, el matrimonio por una línea horizontal, un niño por una línea vertical, varios niños por una ramificación horizontal de una línea vertical (en la forma de la letra P). Lea más sobre estas designaciones
aquí .
Como escribimos anteriormente, los coeficientes de parentesco no pueden caracterizar inequívocamente el parentesco de uno u otro orden: se obtienen los mismos coeficientes de parentesco al comparar pares padre-hijo y hermano-hermana (parentesco de primer orden). Si no es posible establecer la naturaleza de la relación, indique cualquiera de las posibles. Tenga en cuenta que las muestras en el conjunto de datos de prueba tienen identificadores diferentes de los utilizados en el conjunto de datos de entrenamiento.
Las respuestas
deben enviarse a
wgs@atlas.ru mail hasta el 26 de diciembre a las 23:59. Pronto se publicarán dos tareas más, y los resultados finales de las tareas aparecerán el 28 de diciembre. El ganador recibirá la prueba completa del genoma, y el segundo y tercer lugar recibirán la prueba genética Atlas. También habrá premios especiales de
Yandex.Cloud . Los empleados anteriores y actuales de Atlas no participan en la competencia;)