Hoy publicamos la última tarea del ciclo en el que decimos cómo trabajar con datos genéticos.
La primera y la
segunda tarea ya están publicadas: se pueden resolver y enviarnos respuestas. Te advertimos que esta tarea lleva más tiempo que el resto.
El premio principal es el
Genoma Completo .

Anteriormente hemos compartido información útil y enlaces que pueden ser útiles para trabajar con datos bioinformáticos. Le recomendamos que lea primero los artículos anteriores si se los perdió:
¿Qué es el genoma completo y por qué es necesario?Tarea número 1. Descubra el género y el grado de relación.Tarea número 2. Determinación de la estructura de la población.Descargo de responsabilidadEl trabajo con datos genéticos se lleva a cabo en sistemas Unix (Linux, macOS), ya que algunos comandos y software no están disponibles en Windows. Por lo tanto, para los usuarios de Windows, una de las soluciones más simples es alquilar una máquina virtual Linux.
Todas las operaciones descritas a continuación se realizan en la línea de comando - terminal. Antes de comenzar, aprenda a trabajar en una terminal que ejecute su sistema operativo y use comandos, ya que algunos de ellos pueden dañar el sistema operativo y sus datos.
Software requerido
Hemos recopilado la
imagen de una máquina virtual (VM) con todo el software necesario en Yandex.Cloud. Las instrucciones para configurar una VM e instalar software se pueden encontrar en el
artículo con la primera tarea. También hay instrucciones sobre cómo configurar la máquina para usarla de forma gratuita hasta el 31 de diciembre de 2019.
En esta tarea, debe convertir los datos de genotipado del formato VCF al formato 23andMe, cargar los archivos recibidos en el servicio Promethease y familiarizarse con el contenido del informe para cada muestra.
El formato 23andMe es un formato de texto para almacenar datos de genotipado y contiene 4 campos separados por pestañas. El primer campo contiene el identificador de variación (por ejemplo, rsID), el segundo contiene el cromosoma (los valores válidos para este campo son 1-22, X, Y y MT), el tercero contiene la posición en el cromosoma, el cuarto contiene el genotipo (diploide en presencia de dos cromosomas homólogos, haploide en otros casos). Este formato es compatible con muchos servicios de interpretación, por lo que en la tarea trabajaremos con él.
Para completar la tarea, necesita el paquete de software BCFtools. Si aún no lo ha instalado, lea el
artículo con la primera tarea. Contiene instrucciones de instalación. Le recordamos que para participar en la competencia de Año Nuevo 2019, todas las tareas deben completarse.
Además de BCFtools, necesitará
create_23andme.sh el
create_23andme.sh , un script bash que se utiliza para generar datos en formato 23andMe. Este archivo se encuentra en el directorio
/Technical en Yandex.Cloud, así como en el archivo para descargar, disponible a través del enlace en el
artículo .
Toma nota
Existen muchos servicios que analizan los datos de genotipado: MyHeritage, Promethease, FamilyTreeDNA, DNA.LAND, GEDmatch. Proporcionan la descarga de datos de genotipado en varios formatos, a menudo específicos de un proveedor de genotipado particular (Ancestry, 23andMe, MyHeritage, FamilyTreeDNA, GenesForGood y otros). Promethease es el más fiel al formato de datos: puede descargar archivos VCF y 23andMe en este servicio.
Existen varios problemas de compatibilidad entre formatos y servicios:
- Diferentes compañías usan diferentes versiones del genoma para mapear las variaciones genéticas. Este problema se resuelve mediante el llamado Liftover, cuando las posiciones de las variaciones genéticas en los datos fuente se reemplazan por las correspondientes en otra versión del genoma. Por ejemplo, Atlas proporciona datos de genotipado para la versión del genoma GRCh38, y GEDmatch recibe datos de la versión anterior del genoma GRCh37. La conversión de las coordenadas de variaciones genéticas de GRCh38 a GRCh37 se llama elevador.
- Uso de identificadores únicos para variaciones genéticas que no sean rsID. Dichas incompatibilidades se resuelven excluyendo dichas entradas del archivo o anotándolas mediante la asignación de un rsID. El segundo no siempre es posible.
- Los servicios utilizan un conjunto fijo de variaciones genéticas. A veces, una falta de coincidencia de al menos parte de los datos que se descargan da como resultado un error de carga. Este problema es relevante, por ejemplo, para MyHeritage. Se puede resolver resaltando un conjunto de identificadores de variaciones genéticas que no causan un error de carga.
Datos utilizados
Le recordamos que este manual utiliza datos abiertos especialmente seleccionados del proyecto
1000 Genomes . Para el análisis, seleccionamos 10 muestras con información de genotipo de ~ 85 millones de variaciones, que se obtuvieron analizando datos NGS alineados con la versión del genoma GRCh37. Las relaciones familiares y las poblaciones de estas muestras se muestran en la Figura 1.
 Figura 1
Figura 1 El pedigrí de las muestras utilizadas en el VCF (el cuadrado corresponde al género masculino, el círculo al femenino). La línea discontinua corresponde a una relación de segundo orden indeterminada.
Conversión VCF
A continuación se presentan instrucciones para convertir un archivo VCF y cargar los datos recibidos en el servicio Promethease, que recientemente se convirtió en gratuito. Sugerimos que se familiarice con el informe Promethease recibido en cualquiera de las muestras. Use el archivo VCF filtrado por la lista de variaciones obtenidas en la
tarea No. 1 .
El
bcftools query permite extraer cualquier información disponible en un formato especificado por el usuario después del indicador
-f de un archivo VCF. El
HG00731 -s indica el identificador de la muestra (
HG00731 ) para el que extraer datos. El indicador -e se usa para indicar criterios de exclusión, en este caso
'%ID=="."' Excluye entradas que no tienen un rsID. El resultado de la
bcftools query se pasa al script
create_23andme.sh , que convierte los datos al formato TSV con 4 columnas (rsID, cromosoma, posición, genotipo) y los escribe en un archivo. Puede descargar y guardar el script
create_23andme.sh para trabajar con sus propios datos de secuenciación del genoma completo.
El script
create_23andme.sh usa los
create_23andme.sh extraídos del archivo VCF para determinar el tipo de variación genética (variación de un solo nucleótido de SNV, inserción de INS o eliminación de DEL) y escribe el identificador rsID, cromosoma, posición y alelos en
stdout según el tipo específico de variación (A, G, T y C son alelos válidos para el tipo SNV, I y D son designaciones de alelos válidas para los tipos INS y DEL).
Tenga en cuenta que el proceso de conversión lleva mucho tiempo: aproximadamente 4 horas por archivo para una muestra con ~ 1 millón de variaciones. Concurrencia BCFtools no es compatible.
Vaya a
promethease.com y regístrese. Haga clic en el botón Cargar datos sin procesar (Figura 2) y cargue el archivo
HG00731.subset.23andme.txt . Una vez completada la descarga, haga clic en el botón Crear informe gratuito e ingrese el nombre deseado del informe que se generará de acuerdo con sus datos. Después de redactar el informe, recibirá una notificación por correo electrónico y podrá familiarizarse con el contenido del informe. En los informes para cada muestra, encuentre el grupo sanguíneo determinado por el sistema de interpretación de Promethease en el sistema AB0 / Rh (factor Rh - Rh). Verifique que sus resultados cumplan con la Tabla 1.
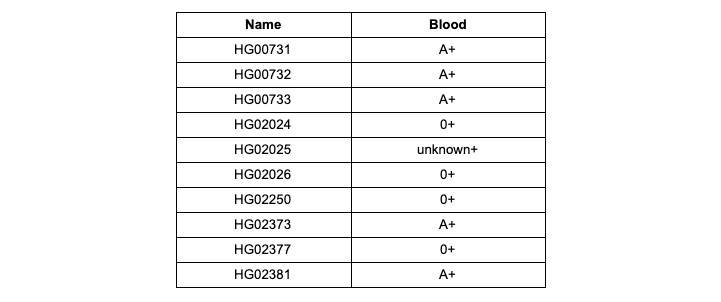 Tabla 1
Tabla 1 . Grupos sanguíneos y factor Rh obtenidos de un análisis de Promethease de muestras de un conjunto de datos de demostración
El atlas utiliza umbrales que difieren de Promethease para incluir un atributo particular en la interpretación por nivel de evidencia. El nivel de evidencia se refiere a la totalidad de los resultados de las pruebas estadísticas y los criterios para la importancia de cada relación observada entre la variación genética y cualquier característica del cuerpo humano. Muchos de los rasgos que se pueden encontrar en el informe Promethease tienen un bajo nivel de evidencia y / o un alto nivel solo en un conjunto limitado de poblaciones, por ejemplo, solo para representantes de la población asiática.
NotaEmpíricamente, hemos instalado una lista de variaciones genéticas basadas en el
chip Infinium Global Screening Array v2.0 que puede cargarse en MyHeritage. Esta lista (
external_interpretation_rsids.txt ) se almacena en un archivo separado en el directorio
/Technical , y se puede usar para filtrar VCF con la conversión posterior por analogía con las instrucciones anteriores. También puede usar este archivo para filtrar los datos de genotipado de un chip para poder cargarlo en MyHeritage. Si ya tiene la prueba genética Atlas, puede cargar los datos de genotipado en un formato desde su cuenta personal y filtrarlos según la lista de variaciones propuesta: la primera columna de los datos cargados desde su cuenta personal.
Tenga en cuenta que los archivos utilizados en este manual siempre contienen un campo ALT relleno (alelo alternativo), lo que permite comprender a qué tipo pertenece cada variación (INS, DEL, SNV) y crear correctamente una entrada en el formato 23andMe. Los datos de secuenciación de todo el genoma en el Atlas contienen el alelo ALT lleno solo en los lugares donde se detectó este alelo; de lo contrario, la información para llenar el campo ALT al generar un archivo VCF simplemente no existe. La salida de datos en sitios de referencia homocigotos (posiciones en el genoma donde no se encontró el alelo de referencia) es necesaria, ya que no solo las variaciones detectadas en la secuencia de nucleótidos, sino también su ausencia tienen un efecto clínico.
La ausencia del alelo ALT en tales posiciones del genoma no nos permite determinar el tipo de variación genética para la cual solo se encontró el alelo de referencia (REF). El registro de genotipos para tales casos se complica por la necesidad de utilizar una fuente de información sobre posibles alelos para esta variación y no está cubierto por esta guía. Si potencialmente utilizará este manual y el script
create_23andme.sh para convertir un archivo VCF obtenido después de una secuenciación de genoma completo a Atlas, el archivo convertido no contendrá genotipos homocigotos de referencia, ya que el script
create_23andme.sh filtra explícitamente dichos registros para eliminar errores al crear registros para inserciones y eliminaciones.
Para que el script
create_23andme.sh siga produciendo genotipos homocigóticos de referencia, debe reemplazar el contenido de las líneas 25–28 en él
... if [ "$ALT" == "." ] || [[ "$ALT" == *"*"* ]] then continue fi ...
en
... if [[ "$ALT" == *"*"* ]] then continue fi if [ "$ALT" == "." ] then echo -e "$RSID\t$CHR\t$POS\t$REF$REF" fi ...
Esta sustitución permite que se muestren entradas
stdout con genotipos de referencia homocigotos. Debe tenerse en cuenta que dichas entradas para inserciones y eliminaciones serán incorrectas, ya que los alelos válidos en el formato utilizado para las inserciones y eliminaciones son I y D, y el script utilizará los alelos A, G, T o C. Para generar correctamente datos para inserciones y deleciones, es necesario saber de antemano qué tipo de variación es característica de una posición dada del genoma en la que no se detectó el alelo ALT. Esta información se puede obtener analizando el alelo ALT si está disponible (ya implementado en
create_23andme.sh ) o utilizando una base de datos externa, por ejemplo, dbSNP (no en
create_23andme.sh ).
Para obtener un informe de Promethease en un archivo VCF completo de secuenciación completa del genoma en Atlas, puede cargar el archivo VCF en Promethease, sin embargo, debe tener en cuenta que el tamaño del archivo comprimido Atlas VCF es de aproximadamente 8 gigabytes, mientras que Promethease le permite cargar archivos no más de 4 gigabytes La descripción de las soluciones a este problema está disponible
aquí . Otra solución es dividir el archivo VCF en varias partes (menos de 4 gigabytes cada una) y cargar cada una como un archivo adicional en el menú de descarga de datos de Promethease.
La tercera tarea de la competencia.
Descargue los datos convertidos de cada una de las 12 muestras del conjunto de datos de prueba, que filtró de acuerdo con la lista de variaciones en la primera tarea, en Promethease y compile una tabla de correspondencia para el identificador de muestra: grupo sanguíneo AB0 / Rh definido por el sistema de interpretación de Promethease (factor Rh - Rh). Los grupos sanguíneos identificados probabilísticamente y registrados con el prefijo "prob" en el informe Promethease, escriben sin el prefijo. Registre valores indefinidos como desconocidos (el factor Rhesus para grupos sanguíneos desconocidos aún debe escribirse, si está definido). Un ejemplo se presenta en la Tabla 1.
La conversión de VCF al formato utilizado anteriormente en la implementación propuesta se simplifica enormemente, pero requiere una cantidad de tiempo considerable. Para la optimización, puede escribir una secuencia de comandos con un bucle que generará automáticamente estos datos, iterando sobre un conjunto de identificadores. Es posible hacer varios scripts de este tipo y transferir cada uno diferentes conjuntos de identificadores de muestra para la ejecución en paralelo, sin embargo, el número de scripts en ejecución paralelos no debe exceder el número de CPU de su computadora / máquina virtual. Una buena descripción de cómo crear tales bucles está disponible
aquí . Cuando trabaje en Yandex.Cloud, puede, si es necesario, crear otra máquina virtual con una gran cantidad de CPU virtuales, lo que reducirá proporcionalmente el tiempo que lleva completar una tarea.
Esta es la última tarea de nuestro ciclo. Las respuestas
deben enviarse a
wgs@atlas.ru mail hasta el 26 de diciembre a las 23:59. Publicaremos las respuestas correctas y los nombres de los ganadores el 28 de diciembre. El ganador recibirá la prueba completa del genoma, y el segundo y tercer lugar recibirán la prueba genética Atlas. También habrá premios especiales de
Yandex.Cloud . Los empleados anteriores y actuales de Atlas no participan en la competencia;)