ماذا لو كان لديك فكرة عن بروتين بارد وصحي ، وكنت ترغب في الحصول عليه في الواقع؟ على سبيل المثال ، هل ترغب في إنشاء لقاح ضد
H. pylori (مثل
الفريق السلوفيني في iGEM 2008 ) عن طريق إنشاء بروتين هجين يجمع شظايا
E. coli flagellin التي تحفز الاستجابة المناعية مع
H. pylori flagellin المعتادة؟
H. pylori Hybrid Flagellin Design مقدم من الفريق السلوفيني في iGEM 2008من المثير للدهشة أننا قريبون جدًا من إنشاء أي بروتين نريده دون ترك دفتر Jupyter ، وذلك بفضل التطورات الأخيرة في علم الجينوم ، والبيولوجيا الاصطناعية ، ومؤخراً في مختبرات السحابة.
في هذه المقالة ، سأعرض شفرة بيثون من فكرة البروتين إلى تعبيره في خلية بكتيرية ، دون لمس ماصة أو التحدث إلى أي شخص. التكلفة الإجمالية ستكون فقط بضع مئات من الدولارات! باستخدام
مصطلحات فيجايا باندي من A16Z ، وهذا هو علم الأحياء 2.0.
وبشكل أكثر تحديدًا ، في المقالة ، يقوم رمز Python الخاص بمختبر السحابة بما يلي:
- توليف تسلسل الحمض النووي الذي يشفر أي بروتين أريد.
- استنساخ هذا الحمض النووي الاصطناعي في ناقل يمكنه التعبير عنه.
- تحول البكتيريا مع هذا المتجه وتأكيد حدوث التعبير.
بيثون الإعداد
أولاً ، إعدادات Python العامة اللازمة لأي مفكرة Jupyter. نحن نستورد بعض وحدات Python المفيدة وننشئ بعض وظائف الأداة المساعدة ، خاصةً لتصور البيانات.
قانونimport re import json import logging import requests import itertools import numpy as np import seaborn as sns import pandas as pd import matplotlib as mpl import matplotlib.pyplot as plt from io import StringIO from pprint import pprint from Bio.Seq import Seq from Bio.Alphabet import generic_dna from IPython.display import display, Image, HTML, SVG def uprint(astr): print(astr + "\n" + "-"*len(astr)) def show_html(astr): return display(HTML('{}'.format(astr))) def show_svg(astr, w=1000, h=1000): SVG_HEAD = '''<?xml version="1.0" standalone="no"?><!DOCTYPE svg PUBLIC "-//W3C//DTD SVG 1.1//EN" "http://www.w3.org/Graphics/SVG/1.1/DTD/svg11.dtd">''' SVG_START = '''<svg viewBox="0 0 {w:} {h:}" version="1.1" xmlns="http://www.w3.org/2000/svg" xmlns:xlink= "http://www.w3.org/1999/xlink">''' return display(SVG(SVG_HEAD + SVG_START.format(w=w, h=h) + astr + '</svg>')) def table_print(rows, header=True): html = ["<table>"] html_row = "</td><td>".join(k for k in rows[0]) html.append("<tr style='font-weight:{}'><td>{}</td></tr>".format('bold' if header is True else 'normal', html_row)) for row in rows[1:]: html_row = "</td><td>".join(row) html.append("<tr style='font-family:monospace;'><td>{:}</td></tr>".format(html_row)) html.append("</table>") show_html(''.join(html)) def clean_seq(dna): dna = re.sub("\s","",dna) assert all(nt in "ACGTN" for nt in dna) return Seq(dna, generic_dna) def clean_aas(aas): aas = re.sub("\s","",aas) assert all(aa in "ACDEFGHIKLMNPQRSTVWY*" for aa in aas) return aas def Images(images, header=None, width="100%"):
مختبرات السحابة
مثل AWS أو أي سحابة حسابية ، يحتوي مختبر السحابة على معدات البيولوجيا الجزيئية ، وكذلك الروبوتات التي تستأجرها عبر الإنترنت. يمكنك إصدار تعليمات لبرامج الروبوت الخاصة بك عن طريق النقر على بعض الأزرار على الواجهة أو عن طريق كتابة التعليمات البرمجية التي تقوم ببرمجتها بنفسك. ليس من الضروري أن تكتب بروتوكولاتك الخاصة ، كما سأفعل هنا ، فإن جزءًا مهمًا من البيولوجيا الجزيئية هو مهام روتينية معيارية ، لذلك من الأفضل عادة الاعتماد على بروتوكول أجنبي موثوق يُظهر تفاعلًا جيدًا مع الروبوتات.
في الآونة الأخيرة ، ظهر عدد من الشركات التي لديها مختبرات سحابية:
Transcriptic ، و
Autodesk Wet Lab Accelerator (beta ، وبنيت على أساس Transcriptic) ،
Arcturus BioCloud (beta) ،
Emerald Cloud Lab (beta) ،
Synthego (لم تبدأ بعد). هناك حتى شركات مبنية على أعلى المعامل السحابية مثل
Desktop Genetics ، والتي تتخصص في كريسبر.
بدأت المقالات العلمية حول استخدام مختبرات السحابة في العلوم الحقيقية في الظهور.
في وقت كتابة هذا التقرير ، كان فقط Transcriptic في المجال العام ، لذلك سنستخدمه. كما أفهمها ، فإن معظم أعمال Transcriptic مبنية على أتمتة البروتوكولات الشائعة ، وكتابة بروتوكولاتك الخاصة في Python (كما سأفعل في هذا المقال) أقل شيوعًا.
 "خلية عمل" نصية مع وجود ثلاجات في القاع ومعدات مخبرية مختلفة على الحامل
"خلية عمل" نصية مع وجود ثلاجات في القاع ومعدات مخبرية مختلفة على الحاملسأقدم تعليمات الروبوتات Transcriptic على
بروتوكول لصناعة السيارات . Autoprotocol هي لغة قائمة على JSON لكتابة بروتوكولات الروبوتات المعملية (والبشر ، كما كانت). يتم صنع Autoprotocol بشكل رئيسي في
مكتبة Python هذه . تم إنشاء اللغة في الأصل ولا تزال مدعومة بواسطة Transcriptic ، لكنها ، كما أفهمها ، مفتوحة تمامًا. هناك
وثائق جيدة.
تتمثل إحدى الأفكار المثيرة للاهتمام في أنه يمكنك كتابة تعليمات للأشخاص في المختبرات البعيدة في الصين أو الهند ، على سبيل المثال ، حول البروتوكول التلقائي ويحتمل أن تحصل على بعض المزايا من استخدام كل من الأشخاص (حكمهم) والروبوتات (عدم الحكم). نحتاج إلى ذكر
protocols.io هنا ، هذه محاولة لتوحيد البروتوكولات لتحسين التكاثر ، ولكن بالنسبة للبشر ، وليس البشر الآليين.
"instructions": [ { "to": [ { "well": "water/0", "volume": "500.0:microliter" } ], "op": "provision", "resource_id": "rs17gmh5wafm5p" }, ... ]
المثال التلقائي للجزء الآليإعدادات بايثون للبيولوجيا الجزيئية
بالإضافة إلى استيراد مكتبات قياسية ، سأحتاج إلى بعض الأدوات البيولوجية الجزيئية المحددة. هذا الرمز هو أساسا للبروتوكول لصناعة السيارات ونسخة.
غالبًا ما يوجد مفهوم "الحجم الميت" في الكود. هذا يعني أن آخر قطرة من السائل لا يمكن أن تأخذها الروبوتات Transcriptic باستخدام ماصة من الأنابيب (لأنها لا تستطيع رؤيتها!). عليك أن تقضي الكثير من الوقت للتأكد من أن القوارير تحتوي على مواد كافية.
قانون import autoprotocol from autoprotocol import Unit from autoprotocol.container import Container from autoprotocol.protocol import Protocol from autoprotocol.protocol import Ref
تخليق الحمض النووي والبيولوجيا الاصطناعية
على الرغم من ارتباطه بالبيولوجيا الاصطناعية الحديثة ، فإن تخليق الحمض النووي تقنية قديمة إلى حد ما. لعقود من الزمان ، تمكنا من إنتاج قليل النوكليوتيدات (أي ، تسلسل الحمض النووي يصل إلى 200 قاعدة). ومع ذلك ، كان دائما مكلفا ، والكيمياء لم تسمح أبدا تسلسل الحمض النووي طويلة. في الآونة الأخيرة ، أصبح من الممكن بسعر معقول تجميع جينات كاملة (حتى آلاف القواعد). هذا الإنجاز يفتح حقًا عصر "البيولوجيا التركيبية".
لقد دفعت
الجينوميات الاصطناعية من كريغ فنتر البيولوجيا التخليقية إلى أبعد ما يكون من خلال
توليف كائن كامل - طوله أكثر من مليون قاعدة. مع زيادة طول الحمض النووي ، لم تعد المشكلة تخليق ، ولكن التجميع (على سبيل المثال ، تجميع تسلسل الحمض النووي توليفها معا). مع كل مجموعة ، يمكنك مضاعفة طول الحمض النووي (أو أكثر) ، لذلك بعد عشرة أو أكثر من التكرار ، تحصل على
جزيء طويل إلى حد ما ! يجب أن يصبح التمييز بين التوليف والتجميع واضحًا للمستخدم النهائي.
قانون مور؟
ينخفض سعر تخليق الحمض النووي بسرعة كبيرة ، من أكثر من 0.30 دولار في العام الماضي إلى ما يقرب من 0.10 دولار اليوم ، لكنه يتطور أكثر مثل البكتيريا من المعالجات. في المقابل ، انخفضت أسعار تسلسل الحمض النووي بشكل أسرع من قانون مور. يتم تعيين هدف
قدره 0.02 دولار لكل قاعدة كنقطة انعطاف حيث يمكنك استبدال الكثير من التلاعب بالحمض النووي المستهلكة للوقت بتوليف بسيط. على سبيل المثال ، عند هذا السعر ، يمكنك توليف كامل البلازميد 3 كيلو بايت مقابل
60 دولارًا وتخطي مجموعة من البيولوجيا الجزيئية. آمل أن نحقق ذلك في غضون عامين.
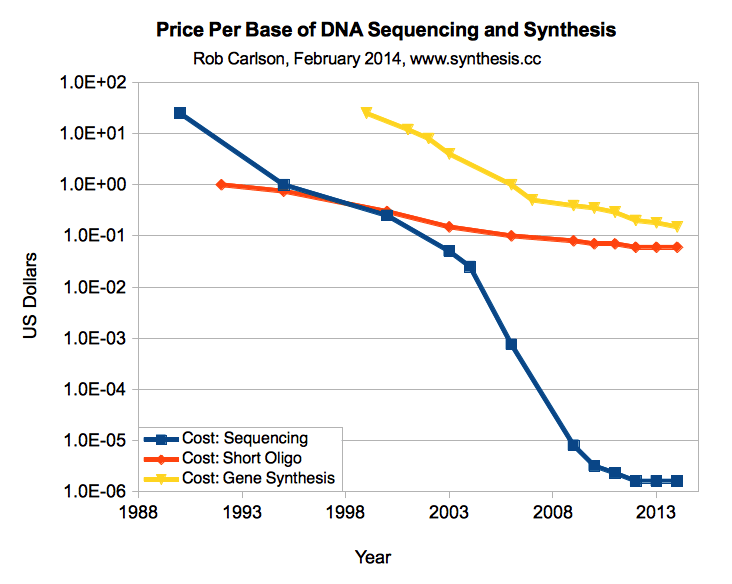 أسعار تركيب الحمض النووي مقارنة بأسعار تسلسل الحمض النووي ، وسعر قاعدة واحدة (كارلسون ، 2014)
أسعار تركيب الحمض النووي مقارنة بأسعار تسلسل الحمض النووي ، وسعر قاعدة واحدة (كارلسون ، 2014)شركات تصنيع الحمض النووي
هناك العديد من الشركات الكبيرة في مجال تخليق الحمض النووي: IDT هي أكبر منتج للأليغنوكليوتيدات ، ويمكنها أيضًا إنتاج "شظايا جينية" أطول (حتى 2
كيلو بايت ) (
gBlocks ).
Gen9 و
Twist و
DNA 2.0 متخصصون عادة في تسلسل الحمض النووي الأطول - هذه هي شركات تصنيع الجينات. هناك أيضًا بعض الشركات الجديدة المثيرة للاهتمام ، مثل
Cambrian Genomics و
Genesis DNA ، التي تعمل على طرق التوليف من الجيل التالي.
تستخدم شركات أخرى ، مثل
Amyris و
Zymergen و
Ginkgo Bioworks ، الحمض النووي الذي توليفه هذه الشركات للعمل على مستوى الجسم. تقوم
الجينوميات الاصطناعية بهذا أيضًا ، ولكنها توليف الحمض النووي نفسه.
أبرمت الجنكة مؤخرًا
صفقة مع Twist لإنشاء 100 مليون قاعدة: أكبر صفقة رأيتها. هذا يثبت أننا نعيش في المستقبل ، حتى أن Twist أعلنت عن رمز ترويجي على Twitter: عندما تشتري 10 ملايين قاعدة من الحمض النووي (تقريبًا جينوم الخميرة بالكامل!) ، ستحصل على 10 ملايين أخرى مجانًا.
 عرض تويتر تويست المتخصصة
عرض تويتر تويست المتخصصةالجزء الأول: تصميم التجربة
البروتين الفلوري الأخضر
في هذه التجربة ، نقوم بتركيب تسلسل الحمض النووي
لبروتين فلوري أخضر بسيط (GFP). تم العثور على بروتين GFP لأول مرة في
قنديل البحر الذي يتألق تحت ضوء الأشعة فوق البنفسجية. هذا بروتين مفيد للغاية لأنه من السهل اكتشاف تعبيره ببساطة عن طريق قياس التألق. توجد خيارات GFP تنتج ألوانًا صفراء وحمراء وبرتقالية وألوانًا أخرى.
من المثير أن نرى كيف تؤثر الطفرات المختلفة على لون البروتين ، وهذه مشكلة تعلم الآلة المحتملة. في الآونة الأخيرة ، سيتعين عليك قضاء الكثير من الوقت في المختبر من أجل هذا ، لكن الآن سأظهر لك أنه (تقريبًا) سهل مثل تحرير ملف نصي!
من الناحية الفنية ، فإن GFP الخاص بي هو خيار المجلد الفائق (sfGFP) مع بعض الطفرات لتحسين الجودة.
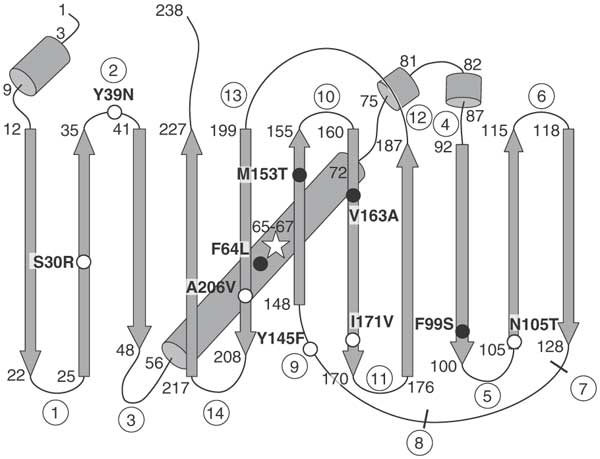 في superfolder-GFP (sfGFP) ، تمنحها بعض الطفرات بعض الخصائص المفيدة.
في superfolder-GFP (sfGFP) ، تمنحها بعض الطفرات بعض الخصائص المفيدة.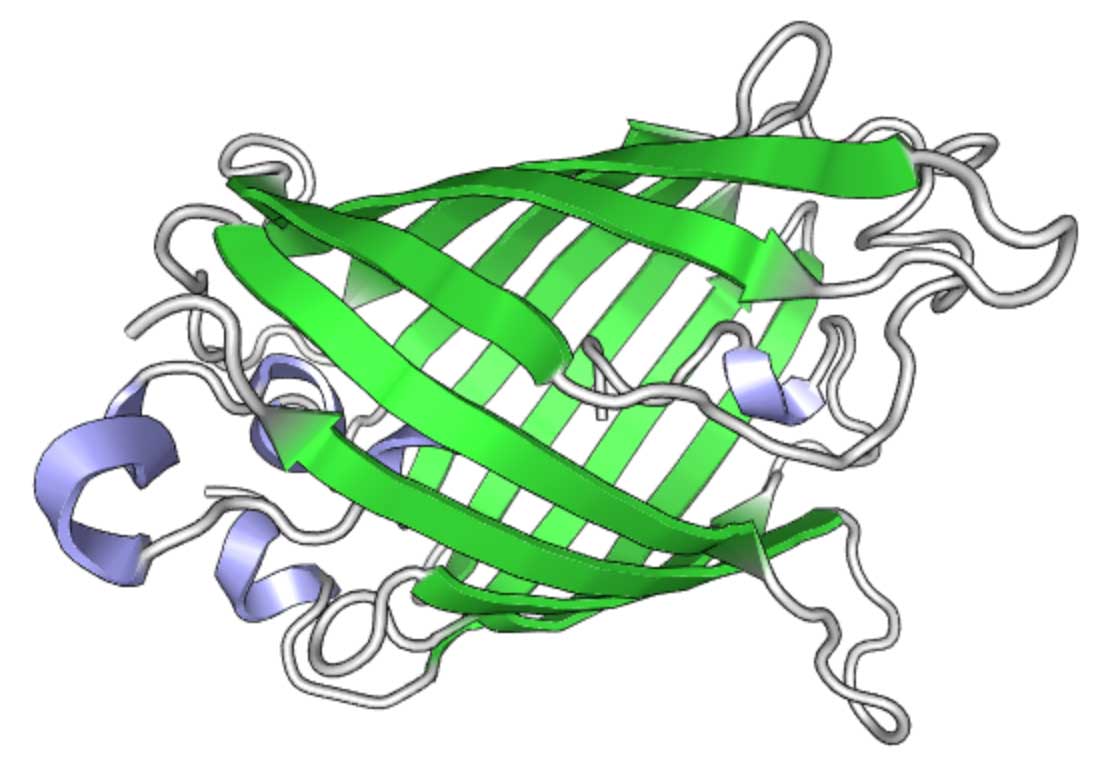 هيكل GFP (تصور باستخدام الكهروضوئية )
هيكل GFP (تصور باستخدام الكهروضوئية )توليف GFP في تويست
كنت محظوظًا بما يكفي للدخول في برنامج اختبار ألفا في تويست ، لذلك استخدمت خدمة تركيب الحمض النووي الخاصة بهم (تفضلوا بوضع طلبي الصغير - شكرًا ، تويست!). هذه شركة جديدة في مجالنا ، مع عملية تخليق مبسطة جديدة. أسعارها حوالي
0.10 دولار لكل قاعدة أو أقل ، لكنها لا
تزال في مرحلة تجريبية ، وتم إغلاق برنامج ألفا الذي شاركت فيه. جمع تويست حوالي 150 مليون دولار ، لذلك التكنولوجيا الخاصة بهم حية.
لقد أرسلت تسلسل الحمض النووي الخاص بي إلى Twist كجدول بيانات Excel (لا يوجد API بعد ، لكن أعتقد أنه سيكون قريبًا) ، وأرسلوا الحمض النووي المركب مباشرةً إلى صندوقي في مختبر Transcriptic (كما أنني استخدمت IDT للتوليف ، لكنها لم ترسل الحمض النووي الحق في النسخ ، الذي يفسد المرح قليلا).
من الواضح أن هذه العملية لم تصبح بعد حالة استخدام نموذجية وتتطلب بعض الدعم ، لكنها نجحت ، بحيث يظل خط الأنابيب بأكمله ظاهريًا. وبدون ذلك ، ربما أحتاج إلى الوصول إلى المختبر - لن ترسل العديد من الشركات الحمض النووي أو الكواشف إلى عنوان منزلها.
 GFP غير ضار ، لذلك يتم تمييز أي نوع
GFP غير ضار ، لذلك يتم تمييز أي نوعناقل البلازميد
للتعبير عن هذا البروتين في البكتيريا ، يحتاج الجين إلى العيش في مكان ما ، وإلا فإن الحمض النووي المشفر للجين يتحلل فورًا. وكقاعدة عامة ، في البيولوجيا الجزيئية ، نستخدم البلازميد ، وهو جزء من الدنا المستدير الذي يعيش خارج الجينوم البكتيري ويعبر عن البروتينات. تعتبر البلازميدات طريقة مناسبة للبكتيريا لتبادل وحدات وظيفية مفيدة قائمة بذاتها ، مثل مقاومة المضادات الحيوية. يمكن أن يكون هناك المئات من البلازميدات في الخلية.
المصطلحات المستخدمة على نطاق واسع هي أن البلازميد هو
ناقل ، وأن الحمض النووي التخليقي عبارة عن إدخال (إدخال). لذا ، نحاول هنا استنساخ الإدخال في ناقل ، ثم تحويل البكتيريا باستخدام المتجه.
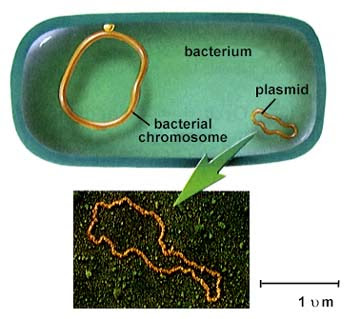 الجينوم البكتيري والبلازميد (وليس على مقياس!) ( ويكيبيديا )
الجينوم البكتيري والبلازميد (وليس على مقياس!) ( ويكيبيديا )pUC19
اخترت البلازميد القياسي إلى حد ما في سلسلة
pUC19 . غالبًا ما يستخدم هذا البلازميد ، ولأنه متاح كجزء من قائمة الجرد القياسية ، فإننا لسنا بحاجة لإرسال أي شيء إليهم.
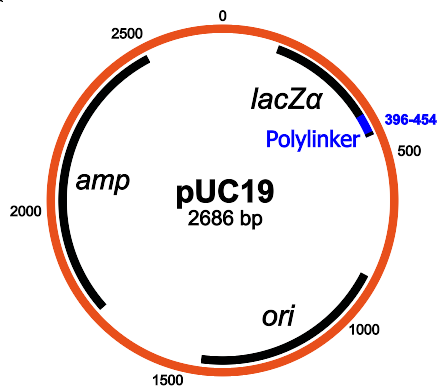 هيكل pUC19: المكونات الرئيسية هي جين مقاومة الأمبيسلين ، lacZα ، MCS / polylinker ، وأصل النسخ المتماثل (ويكيبيديا)
هيكل pUC19: المكونات الرئيسية هي جين مقاومة الأمبيسلين ، lacZα ، MCS / polylinker ، وأصل النسخ المتماثل (ويكيبيديا)يتميز PUC19 بوظيفة لطيفة: نظرًا لأنه يحتوي على جين lacZα ، فيمكنك استخدام طريقة
التحديد الزرقاء والبيضاء لمعرفة أي المستعمرات نجحت عملية الإدراج. هناك حاجة إلى مادتين كيميائيتين:
IPTG و
X-gal ، وتعمل الدائرة على النحو التالي:
- IPTG يستحث التعبير lacZα.
- إذا تم إلغاء تنشيط lacZα عبر إدخال DNA في موقع الاستنساخ المتعدد ( MCS / polylinker ) في lacZα ، فلن يتمكن البلازميد من تحلل X-gal وستكون هذه المستعمرات بيضاء بدلاً من اللون الأزرق.
- لذلك ، يؤدي الإدراج الناجح إلى إنتاج مستعمرات بيضاء ، والإنتاج الفاشل ينتج مستعمرات زرقاء.
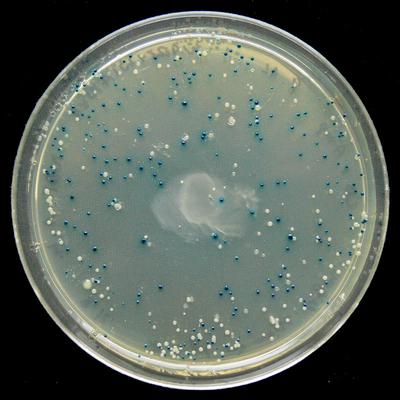 يظهر التحديد الأزرق والأبيض حيث تم إلغاء تنشيط تعبير lacZα ( ويكيبيديا )وثائق openwetware
يظهر التحديد الأزرق والأبيض حيث تم إلغاء تنشيط تعبير lacZα ( ويكيبيديا )وثائق openwetware تقول:
لا يحتاج E. coli DH5α إلى IPTG للحث على التعبير من مروج lac ، حتى إذا تم التعبير عن مثبط Lac في الضغط. يتجاوز عدد النسخ لمعظم البلازميدات عدد المكثفات في الخلايا. إذا كنت بحاجة إلى أقصى قدر من التعبير ، فأضف IPTG إلى تركيز نهائي يبلغ 1 مم.
تسلسل الحمض النووي الاصطناعية
SfGFP تسلسل الحمض النووي
من السهل الحصول على تسلسل الحمض النووي لـ sfGFP من خلال أخذ
تسلسل البروتين وترميزه
باستخدام الكودونات المناسبة للكائن الحي المضيف (هنا ،
E. coli ). هذا بروتين متوسط الحجم يحتوي على 236 من الأحماض الأمينية ، وبالتالي فإن تكاثر الحمض النووي عند 10 سنتات يكلف حوالي
70 دولارًا لكل قاعدة.
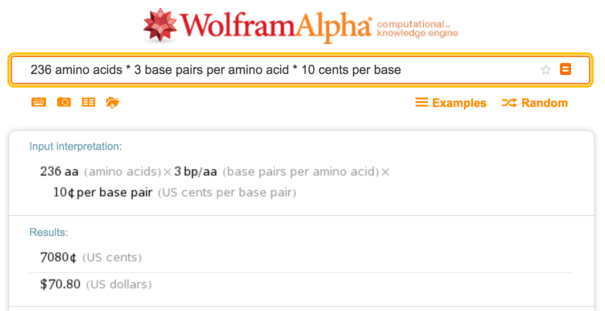 ولفرام ألفا ، حساب تكلفة التوليف
ولفرام ألفا ، حساب تكلفة التوليفأول 12 قاعدة من sfGFP لدينا هي
تسلسل Shine-Delgarno ، الذي أضفته بنفسي ، والذي من الناحية النظرية يجب أن يزيد التعبير (AGGAGGACAGCT ، ثم ATG (
بدء كودون ) يطلق البروتين). وفقًا لأداة حسابية تم تطويرها بواسطة
Salis Lab (
شرائح المحاضرات ) ، يمكننا أن نتوقع تعبيرًا متوسطًا إلى عاليًا لبروتيننا (معدل بدء الترجمة يبلغ 10،000 "وحدة تعسفية").
sfGFP_plus_SD = clean_seq(""" AGGAGGACAGCTATGTCGAAAGGAGAAGAACTGTTTACCGGTGTGGTTCCGATTCTGGTAGAACTGGA TGGGGACGTGAACGGCCATAAATTTAGCGTCCGTGGTGAGGGTGAAGGGGATGCCACAAATGGCAAAC TTACCCTTAAATTCATTTGCACTACCGGCAAGCTGCCGGTCCCTTGGCCGACCTTGGTCACCACACTG ACGTACGGGGTTCAGTGTTTTTCGCGTTATCCAGATCACATGAAACGCCATGACTTCTTCAAAAGCGC CATGCCCGAGGGCTATGTGCAGGAACGTACGATTAGCTTTAAAGATGACGGGACCTACAAAACCCGGG CAGAAGTGAAATTCGAGGGTGATACCCTGGTTAATCGCATTGAACTGAAGGGTATTGATTTCAAGGAA GATGGTAACATTCTCGGTCACAAATTAGAATACAACTTTAACAGTCATAACGTTTATATCACCGCCGA CAAACAGAAAAACGGTATCAAGGCGAATTTCAAAATCCGGCACAACGTGGAGGACGGGAGTGTACAAC TGGCCGACCATTACCAGCAGAACACACCGATCGGCGACGGCCCGGTGCTGCTCCCGGATAATCACTAT TTAAGCACCCAGTCAGTGCTGAGCAAAGATCCGAACGAAAAACGTGACCATATGGTGCTGCTGGAGTT CGTGACCGCCGCGGGCATTACCCATGGAATGGATGAACTGTATAAA""") print("Read in sfGFP plus Shine-Dalgarno: {} bases long".format(len(sfGFP_plus_SD))) sfGFP_aas = clean_aas("""MSKGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLTYG VQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNFNSHNVYITADKQKN GIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHGMDELYK""") assert sfGFP_plus_SD[12:].translate() == sfGFP_aas print("Translation matches protein with accession 532528641")
اقرأ في sfGFP plus Shine-Dalgarno: 726 قواعد طويلة
ترجمة تطابق البروتين مع الانضمام 532528641
PUC19 تسلسل الحمض النووي
أولاً ، أتحقق من أن
تسلسل pUC19 الذي قمت بتنزيله من NEB له الطول الصحيح ويتضمن
polylinker المتوقعة.
pUC19_fasta = !cat puc19fsa.txt pUC19_fwd = clean_seq(''.join(pUC19_fasta[1:])) pUC19_rev = pUC19_fwd.reverse_complement() assert all(nt in "ACGT" for nt in pUC19_fwd) assert len(pUC19_fwd) == 2686 pUC19_MCS = clean_seq("GAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTT") print("Read in pUC19: {} bases long".format(len(pUC19_fwd))) assert pUC19_MCS in pUC19_fwd print("Found MCS/polylinker")
اقرأ في pUC19: 2686 قواعد طويلة
وجدت MCS / polylinker
نقوم ببعض QCs الأساسية للتأكد من وجود EcoRI و BamHI في pUC19 مرة واحدة فقط (تتوفر إنزيمات التقييد التالية في قائمة الجرد بشكل افتراضي:
PstI و
PvuII و
EcoRI و
BamHI و
BbsI و
BsmBI ).
REs = {"EcoRI":"GAATTC", "BamHI":"GGATTC"} for rename, res in REs.items(): assert (pUC19_fwd.find(res) == pUC19_fwd.rfind(res) and pUC19_rev.find(res) == pUC19_rev.rfind(res)) assert (pUC19_fwd.find(res) == -1 or pUC19_rev.find(res) == -1 or pUC19_fwd.find(res) == len(pUC19_fwd) - pUC19_rev.find(res) - len(res)) print("Asserted restriction enzyme sites present only once: {}".format(REs.keys()))
الآن نحن ننظر إلى تسلسل lacZα والتحقق من أنه لا يوجد شيء غير متوقع. على سبيل المثال ، يجب أن تبدأ بـ Met وتنتهي برمز إيقاف. كما أنه من السهل التأكد من أن هذا هو كامل 324 نقطة أساس lacZα ORF عن طريق تحميل تسلسل pUC19 في
عارض snapgene الحرة.
lacZ = pUC19_rev[2217:2541] print("lacZα sequence:\t{}".format(lacZ)) print("r_MCS sequence:\t{}".format(pUC19_MCS.reverse_complement())) lacZ_p = lacZ.translate() assert lacZ_p[0] == "M" and not "*" in lacZ_p[:-1] and lacZ_p[-1] == "*" assert pUC19_MCS.reverse_complement() in lacZ assert pUC19_MCS.reverse_complement() == pUC19_rev[2234:2291] print("Found MCS once in lacZ sequence")
تسلسل lacZ: ATGACCATGATTACGCCAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGTAATAGCGAAGAGGCCCGCACCGATCGCCCTTCCCAACAGTTGCGCAGCCTGAATGGCGAATGGCGCCTGATGCGGTATTTTCTCCTTACGCATCTGTGCGGTATTTCACACCGCATATGGTGCACTCTCAGTACAATCTGCTCTGATGCCGCATAG
تسلسل r_MCS: AAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTC
وجدت MCS مرة واحدة في تسلسل lacZ
الجمعية جيبسون
تجميع الحمض النووي يعني ببساطة شظايا تشابك. عادةً ما تجمع عدة شظايا من الحمض النووي في شريحة أطول ، ثم تنسخها في بلازميد أو جينوم. في هذه التجربة ، أرغب في استنساخ جزء واحد من الحمض النووي في البلازميد pUC19 أسفل مروج lac للتعبير في
E. coli .
هناك العديد من طرق الاستنساخ (مثل
NEB ،
openwetware ،
addgene ). هنا سأستخدم مجموعة جيبسون (
التي طورها دانييل جيبسون في Synthetic Genomics في عام 2009) ، وهي ليست بالضرورة الطريقة الأرخص ، ولكنها بسيطة ومرنة. تحتاج فقط إلى وضع الحمض النووي الذي تريد جمعه (مع التداخل المناسب) في أنبوب اختبار مع Gibson Assembly Master Mix ، وسوف يتجمع نفسه!
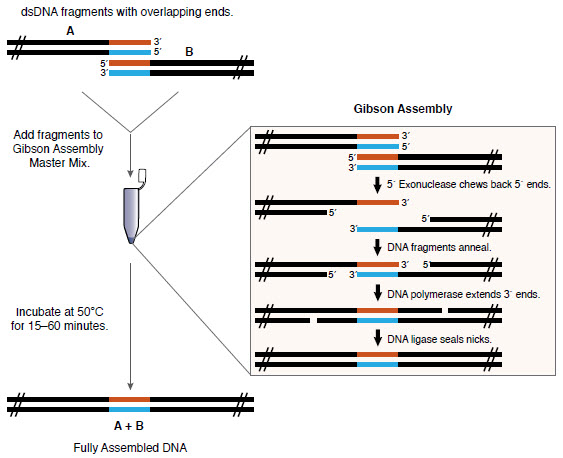 مراجعة جمعية جيبسون ( NEB )
مراجعة جمعية جيبسون ( NEB )مصدر المواد
نبدأ مع 100 نانوغرام من الحمض النووي الاصطناعية في 10 ميكرولتر من السائل. هذا يساوي 0.21 بيكومول من الحمض النووي أو تركيز 10 نانوغرام / ميكرولتر.
pmol_sfgfp = convert_ug_to_pmol(0.1, len(sfGFP_plus_SD)) print("Insert: 100ng of DNA of length {:4d} equals {:.2f} pmol".format(len(sfGFP_plus_SD), pmol_sfgfp))
إدراج: 100ng من الحمض النووي بطول 726 يساوي 0.21 pmol
وفقًا
لبروتوكول تجميع NEB ، هذا مادة مصدر كافية:
يوصي NEB بما مجموعه 0.02-0.5 بيكومول من شظايا الحمض النووي عندما يتم تجميع شظايا 1 أو 2 في ناقل ، أو 0.2-1.0 بيكومول من شظايا الحمض النووي عندما يتم جمع 4-6 شظايا.
0.02-0.5 مساءً * X μl
* كفاءة الاستنساخ الأمثل هو 50-100 نانوغرام من ناقلات مع فائض 2-3 أضعاف من الإدراج. استخدم 5 مرات أكثر من المكونات إذا كان الحجم أقل من 200 نقطة أساس. يجب ألا يتجاوز الحجم الإجمالي لشظايا PCR غير المرشحة في تفاعل مجموعة Gibson 20٪.
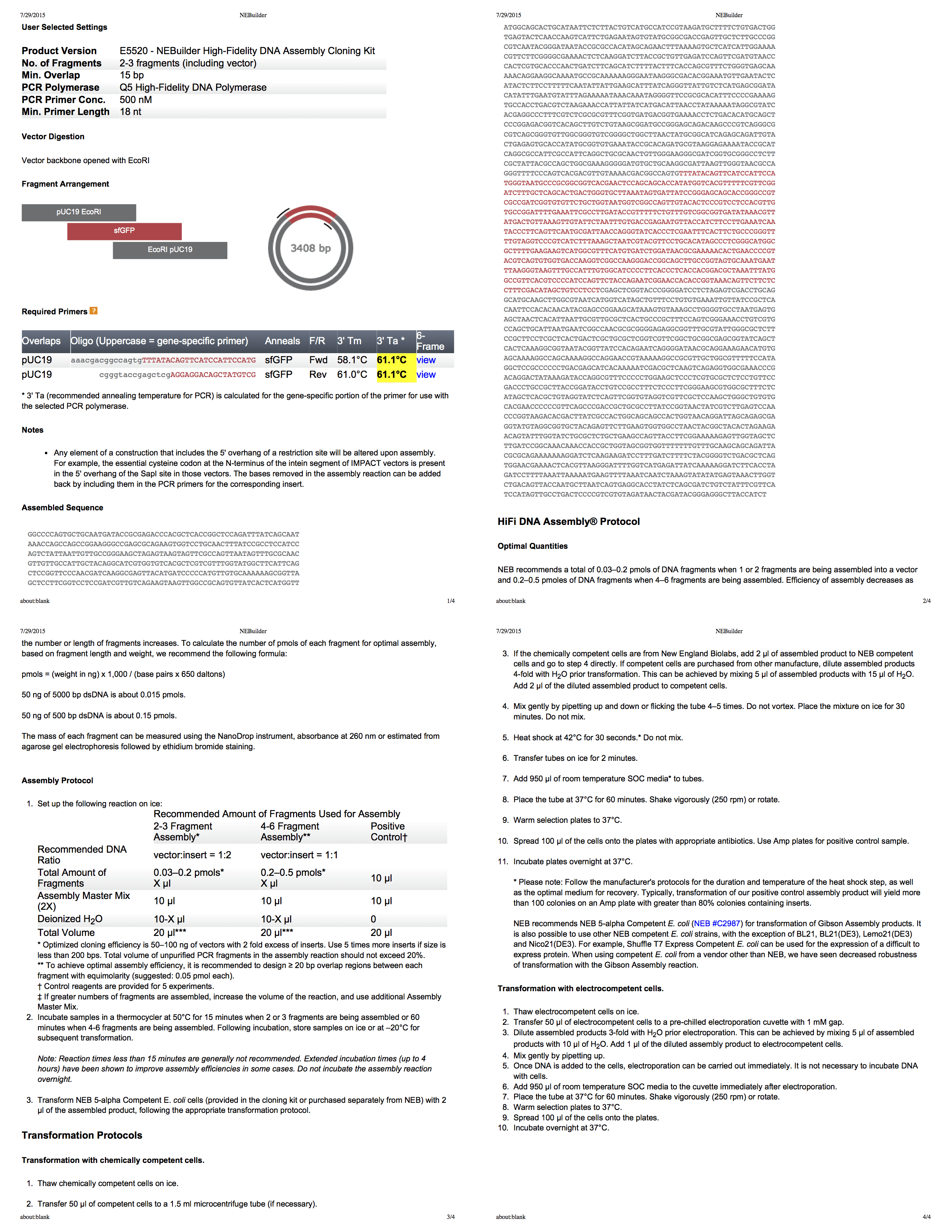
NEBuilder لتجميع جيبسون
Biolab's NEBuilder هي أداة رائعة حقًا لإنشاء بروتوكول إنشاء Gibson. حتى أنه يولد لك PDF شامل من أربع صفحات مع جميع المعلومات. باستخدام هذه الأداة ، نقوم بتطوير بروتوكول لخفض pUC19 باستخدام EcoRI ، ومن ثم استخدام تفاعل PCR [PCR ، يسمح تفاعل البلمرة المتسلسل بتحقيق زيادة كبيرة في التركيزات الصغيرة لشظايا معينة من الحمض النووي في المواد البيولوجية - تقريبًا. لكل.] لإضافة شظايا من الحجم المناسب إلى الإدراج.
الجزء الثاني: التجربة
تتكون التجربة من أربع مراحل:
- تفاعل إدراج سلسلة بوليميريز لإضافة مواد ذات تسلسل جانبي.
- قطع البلازميد لاستيعاب الإدراج.
- تجميع بواسطة جيبسون الإدراج والبلازميدات.
- التحول من البكتيريا باستخدام البلازميد تجميعها.
الخطوة 1. الإدراج PCR
يعتمد تجميع جيبسون على تسلسل الحمض النووي الذي تجمعه ، مع وجود تسلسل متداخل (انظر بروتوكول NEB مع تعليمات مفصلة أعلاه). بالإضافة إلى التضخيم البسيط ، يتيح لك PCR أيضًا إضافة تسلسل الحمض النووي الخاص بالجناح من خلال تضمين تسلسل إضافي في الاشعال (يمكن استنساخه أيضًا
باستخدام OE-PCR فقط ).
نحن توليف الاشعال وفقا لبروتوكول NEB أعلاه. جربت
بروتوكول Quickstart على موقع Transcriptic ، لكن لا يزال
هناك أمر بروتوكول تلقائي . لا يقوم Transcriptic نفسه بتوليف النكليوتيدات ، لذا بعد 1-2 يوم من الانتظار ، تظهر هذه الاشعارات بطريقة سحرية في قائمة مخزوني (لاحظ أن الجزء الخاص بالجينات من المواد الأولية موضح في الحالة العليا أدناه ، لكن هذه مجرد أشياء تجميلية).
insert_primers = ["aaacgacggccagtgTTTATACAGTTCATCCATTCCATG", "cgggtaccgagctcgAGGAGGACAGCTATGTCG"]
تحليل التمهيدي
يمكنك تحليل خصائص هذه الاشعال باستخدام
IDT OligoAnalyzer .
عند تصحيح أخطاء تجربة PCR ، من المفيد معرفة نقاط الانصهار واحتمالية حدوث تأثير جانبي لباطل التمهيدي ، على الرغم من أن بروتوكول NEB سيحدد تقريبًا الاشعال مع خصائص جيدة. جزء محدد من الجينات من الجناح (أحرف كبيرة)
درجة حرارة الذوبان: 51C ، 53.5C
تسلسل كامل
درجة حرارة الذوبان: 64.5C ، 68.5C
دبوس الشعر: -4dG ، -5dG
ديمر الذاتي: -9dG ، -16dG
Heterodimer: -6dG
لقد مررت بالعديد من عمليات تكرار PCR قبل الحصول على نتائج مرضية ، بما في ذلك التجارب مع العديد من العلامات التجارية المختلفة لمزج PCR. نظرًا لأن كل تكرار من هذه التكرارات قد يستغرق عدة أيام (اعتمادًا على طول قائمة الانتظار إلى المختبر) ، فإن الأمر يستحق قضاء بعض الوقت في تصحيح الأخطاء مسبقًا: هذا يوفر الكثير من الوقت على المدى الطويل. مع زيادة قوة المعمل السحابي ، يجب أن تصبح هذه المشكلة أقل حدة. ومع ذلك ، فمن غير المرجح أن ينجح البروتوكول الأول - فهناك العديد من المتغيرات.قانون """ PCR overlap extension of sfGFP according to NEB protocol. v5: Use 3/10ths as much primer as the v4 protocol. v6: more complex touchdown pcr procedure. The Q5 temperature was probably too hot v7: more time at low temperature to allow gene-specific part to anneal v8: correct dNTP concentration, real touchdown """ p = Protocol()
تحذير: الجذر: انخفاض حجم البئر sfGFP 1 / sfGFP 1: 2.0: ميكروليتر
sfGFP 1 / sfGFP 1 2.0: microliter {'dilution': '0.25ng / ul'}
sfgfp_pcroe_v5_puc19_primer1_10uM 75.0: microliter {}
sfgfp_pcroe_v5_puc19_primer2_10uM 75.0: microliter {}
الحجم الموحد 52.0: ميكروليتر
Protocol 1. Amplify the insert (oligos previously synthesized)
---------------------------------------------------------------
✓ Protocol analyzed
11 instructions
8 containers
Total Cost: $32.18
Workcell Time: $4.32
Reagents & Consumables: $27.86 : PCR
في الجل ، يمكنك تقييم الحجم الصحيح للمنتج بعد زيادة التركيز (موضع الشريط في الجل) والكمية الصحيحة (الشريط الداكن). يحتوي الجل على سلم يتوافق مع أطوال وكميات مختلفة من الحمض النووي التي يمكن استخدامها للمقارنة.في صورة الجل أدناه ، تحتوي العصابات D1 و E1 و F1 ، على التوالي ، على 2 ميكرولتر و 4 ميكرولتر و 8 ميكروليتر من المنتج المضخم. يمكنني تقدير كمية الحمض النووي في كل حارة مقارنة مع الحمض النووي في السلم (50 نانوغرام من الحمض النووي لكل حارة في السلم). أعتقد أن النتائج تبدو نظيفة جدا.حاولت استخدام GelEval لتحليل الصور وتقدير التركيزوبنجاح كبير ، على الرغم من أنني لست متأكدًا مما إذا كان هذا أكثر دقة من الطريقة الأكثر سذاجة. ومع ذلك ، أدت التغييرات الطفيفة في موقع وحجم العصابات إلى تغييرات كبيرة في تقدير كمية الحمض النووي. أفضل تقديري لكمية الحمض النووي في المنتج المضخم باستخدام GelEval هو 40 نانوغرام / ميكرولتر.على افتراض أننا محدودون بكمية التمهيدي في الخليط وليس بكمية dNTP أو الإنزيم ، حيث أن لدي 12.5 pmol لكل تمهيدي ، وهذا يعني أن الحد الأقصى النظري هو 6 ميكروغرام من الحمض النووي 740bp في 25 ميكرولتر. نظرًا لأن تقديري للمبلغ الإجمالي للحمض النووي باستخدام GelEval هو 40 نانوغرام × 25 ميكرولتر (1 ميكروغرام أو 2 مساءً) ، فإن هذه النتائج معقولة جدًا وقريبة من ما يجب أن أتوقعه في ظل ظروف مثالية.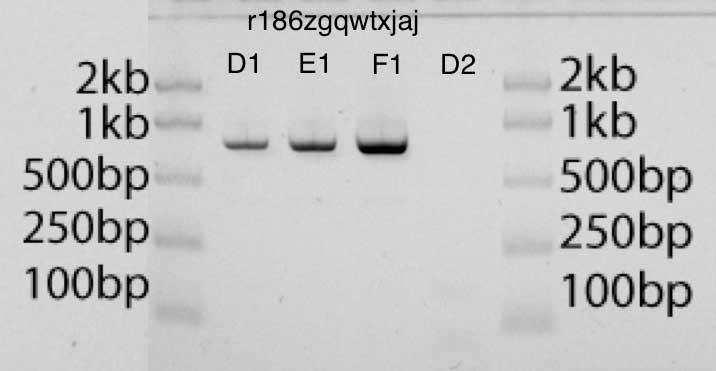 - EcoRI- pUC19, (D1, E1, F1), (D2)
- EcoRI- pUC19, (D1, E1, F1), (D2)PCR
بدأت Transcriptic مؤخرًا في تقديم بيانات تشخيصية مفيدة ومفيدة من الروبوتات الخاصة بها. في وقت كتابة هذا التقرير ، لم تكن متوفرة للتنزيل ، لذلك في الوقت الحالي ليس لدي سوى صورة لدرجات الحرارة أثناء ركوب الدراجات الحراري.تبدو البيانات جيدة ، بدون قمم أو أحجام غير متوقعة. ما مجموعه 35 دورة PCR ، ولكن يتم تنفيذ بعض هذه الدورات في درجات حرارة مرتفعة للغاية كجزء من الهبوط PCR . في محاولاتي السابقة لتضخيم هذا الجزء - الذي كان هناك عدة! - كانت هناك مشاكل مع تهجين الاشعال ، لذلك هنا PCR يعمل لفترة طويلة في درجات حرارة عالية ، والتي ينبغي أن تزيد من الدقة. التشخيصات الحرارية-الدائرية ل PCR touchdown: كتلة ، عينة وتغطية درجات الحرارة لمدة 35 دورة و 42 دقيقة
التشخيصات الحرارية-الدائرية ل PCR touchdown: كتلة ، عينة وتغطية درجات الحرارة لمدة 35 دورة و 42 دقيقةالخطوة 2. قطع البلازميد
لإدخال الحمض النووي sfGFP لدينا في pUC19 ، تحتاج أولاً إلى قطع البلازميد. باتباع بروتوكول NEB ، أفعل ذلك باستخدام إنزيم تقييد EcoRI . هناك الكواشف التي أحتاجها في المخزون النسخي القياسي: هذا هو NEB EcoRI و 10x CutSmart العازلة ، وكذلك NEB pUC19 البلازميد .للحصول على معلومات ، فيما يلي الأسعار من مخزونها. في الواقع ، أنا أدفع جزءًا فقط من السعر ، نظرًا لأن Transcriptic يأخذ المبلغ المستهلك فعليًا: البند معرف المبلغ تركيز السعر
------------ ------ ------------- ----------------- - ----
CutSmart 10x B7204S 5 ml 10 X $ 19.00
EcoRI R3101L 50،000 وحدة / 20،000 وحدة / مل 225.00 دولار
pUC19 N3041L 250 ميكروغرام 1000 ميكروغرام / مل 268.00 دولار
تابعت بروتوكول NEB قدر الإمكان:. 10X dH2O 1X. , , , , . 50 5 10x NEBuffer , dH2O.
, 1 λ 1 37°C 50 . , 5-10 10-20 1- .
1 50 .
قانون """Protocol for cutting pUC19 with EcoRI.""" p = Protocol() experiment_name = "puc19_ecori_v3" options = {} inv = { 'water': "rs17gmh5wafm5p",
Volumes: re_tube:135.0:microliter water_tube:383.0:microliter EcoRI:30.0:microliter
Consolidated volume: 78.0:microliter
✓ Protocol analyzed
12 instructions
5 containers
Total Cost: $30.72
وقت عمل الخلية: 3.38 دولار
الكواشف والمواد الاستهلاكية: 27.34 دولار
النتائج: قطع البلازميد
لقد أجريت هذه التجربة مرتين في ظل ظروف مختلفة قليلاً ومع المواد الهلامية ذات الأحجام المختلفة ، ولكن النتائج متطابقة تقريبًا. أنا أحب كلا المواد الهلامية.في البداية ، لم أخصص مساحة كافية للحجم "الميت" (في أنابيب الاختبار من 1.5 مل ، حجم الميت هو 15 ميكرولتر!). أعتقد أن هذا يفسر الفرق بين D1 و E1 (يجب أن يكون النطاقان متطابقان). من السهل حل مشكلة الحجم الميت عن طريق إنشاء إمداد عمل مناسب لـ EcoRI المخفف في بداية البروتوكول.على الرغم من هذا الخطأ ، يوجد في النطاقين D1 و E1 نطاقات قوية في الموضع الصحيح البالغ 2.6 كيلو بايت. على الشريط D2 ، بلازميد غير مُقطع: كما هو متوقع ، فإنه غير مرئي في أحد الهلام وبالكاد يظهر في آخر.اثنين من صور هلام تبدو مختلفة تماما. هذا يرجع جزئيا إلى حقيقة أن هذه الخطوة نسختها لم تتم بعد.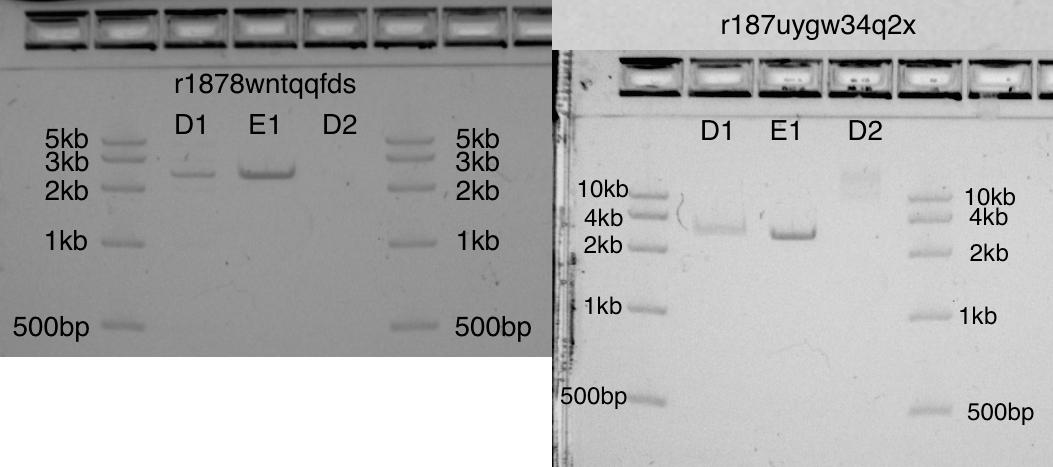 جلان يظهران قطع pUC19 (2.6 كيلو بايت) في النطاقين D1 و E1 ، و pUC19 غير المصقول في D2
جلان يظهران قطع pUC19 (2.6 كيلو بايت) في النطاقين D1 و E1 ، و pUC19 غير المصقول في D2الخطوة 3. الجمعية جيبسون
أسهل طريقة للتحقق ما إذا بلدي بناء من قبل جيبسون - جمع وإدخال البلازميد، ومن ثم استخدام معيار الاشعال وM13 (الذي تطويق الإدراج) لتضخيم DNA البلازميد، والمدرجة تشغيل QPCR و هلام للتأكد من أن التضخيم عمل. يمكنك أيضًا تشغيل رد فعل تسلسلي للتأكد من إدراج كل شيء كما هو متوقع ، لكنني قررت تركه لاحقًا.إذا لم تعمل مجموعة Gibson ، فلن يعمل تضخيم M13 لأنه تم قطع البلازميد بين متسلسلين M13.قانون """Debugging transformation protocol: Gibson assembly followed by qPCR and a gel v2: include v3 Gibson assembly""" p = Protocol() options = {} experiment_name = "debug_sfgfp_puc19_gibson_seq_v2" inv = { "water" : "rs17gmh5wafm5p",
تحذير: الجذر: انخفاض حجم البئر sfgfp_puc19_gibson_v1_clone / sfgfp_puc19_gibson_v1_clone: 11.0: microliter
✓ تحليل البروتوكول
11 تعليمات
6 حاويات
التكلفة الإجمالية: 32.09 دولار
وقت العمل: 6.98 دولار
الكواشف والمواد الاستهلاكية: 25.11 دولار
النتائج: qPCR لتجميع جيبسون
يمكنني الوصول إلى بيانات qPCR بتنسيق JSON من خلال Transcriptic API. هذه الميزة غير موثقة جيدًا ، ولكنها يمكن أن تكون مفيدة للغاية. تتيح لك واجهات برمجة التطبيقات حتى الوصول إلى بعض البيانات التشخيصية من الروبوتات ، والتي يمكن أن تساعد في تصحيح الأخطاء.أولاً ، نطلب بيانات الإطلاق: project_id, run_id = "p16x6gna8f5e9", "r18mj3cz3fku7" api_url = "https://secure.transcriptic.com/hgbrian/{}/runs/{}/data.json".format(project_id, run_id) data_response = requests.get(api_url, headers=tsc_headers) data = data_response.json()
ثم نحدد هذا المعرف للحصول على بيانات "المعالجة اللاحقة" الخاصة بـ qPCR: qpcr_id = data['debug_sfgfp_puc19_gibson_seq_v1_qpcr']['id'] pp_api_url = "https://secure.transcriptic.com/data/{}.json?key=postprocessed_data".format(qpcr_id) data_response = requests.get(pp_api_url, headers=tsc_headers) pp_data = data_response.json()
فيما يلي قيم Ct (حد الدورة) لكل أنبوب. Ct هي ببساطة النقطة التي يتجاوز فيها التألق قيمة معينة. تقول تقريبًا مقدار الحمض النووي الموجود حاليًا (وبالتالي ، حيث بدأنا تقريبًا).
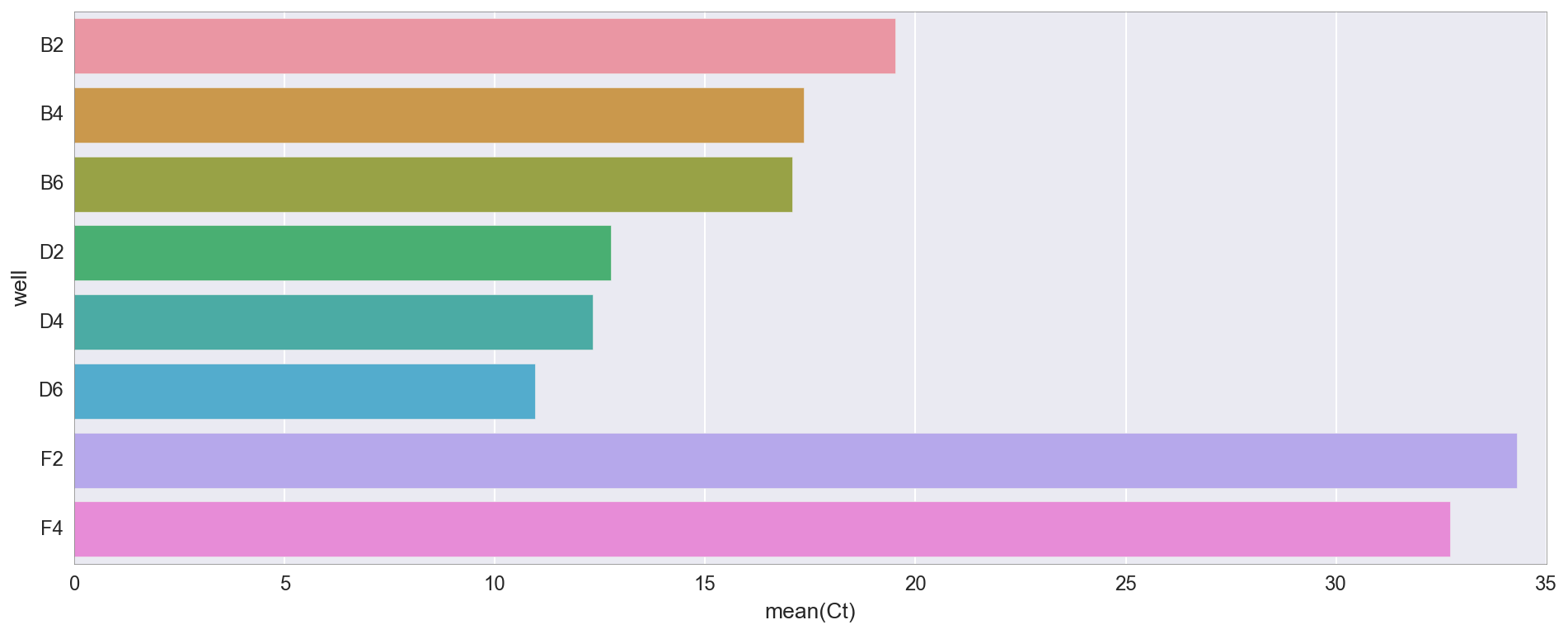 كما ترون ، يحدث التضخيم في المقام الأول في أنابيب الاختبار D2 / 4/6 (حيث يكون الحمض النووي من مجموعة Gibson الخاصة بي هو "v3") ، ثم B2 / 4/6 (تجميع Gibson هو "v1"). الاختلافات بين v1 و v3 هي أن v3 DNA مخفف 4X وفقًا لبروتوكول NEB ، ولكن يجب أن يعمل كلا الخيارين. هناك بعض التضخيم بعد الدورة 30 في أنابيب التحكم (F2 ، F4) تفتقر إلى قالب الحمض النووي ، ولكن هذا ليس من غير المألوف ، لأنها تشمل الكثير من الحمض النووي التمهيدي.يمكنني أيضا رسم منحنى التضخيم qPCR لرؤية ديناميات التضخيم.
كما ترون ، يحدث التضخيم في المقام الأول في أنابيب الاختبار D2 / 4/6 (حيث يكون الحمض النووي من مجموعة Gibson الخاصة بي هو "v3") ، ثم B2 / 4/6 (تجميع Gibson هو "v1"). الاختلافات بين v1 و v3 هي أن v3 DNA مخفف 4X وفقًا لبروتوكول NEB ، ولكن يجب أن يعمل كلا الخيارين. هناك بعض التضخيم بعد الدورة 30 في أنابيب التحكم (F2 ، F4) تفتقر إلى قالب الحمض النووي ، ولكن هذا ليس من غير المألوف ، لأنها تشمل الكثير من الحمض النووي التمهيدي.يمكنني أيضا رسم منحنى التضخيم qPCR لرؤية ديناميات التضخيم. f, ax = plt.subplots(figsize=(16,6)) ax.set_color_cycle(['#fb6a4a', '#de2d26', '#a50f15', '#74c476', '#31a354', '#006d2c', '#08519c', '#6baed6']) amp0 = pp_data['amp0']['SYBR']['baseline_subtracted'] _ = [plt.plot(amp0[w_n[well]], label=well) for well in ['B2', 'B4', 'B6', 'D2', 'D4', 'D6', 'F2', 'F4']] _ = ax.set_ylim(0,) _ = plt.title("qPCR (reds=Gibson v1, greens=Gibson v3, blues=control)") _ = plt.legend(bbox_to_anchor=(1, .75), bbox_transform=plt.gcf().transFigure)
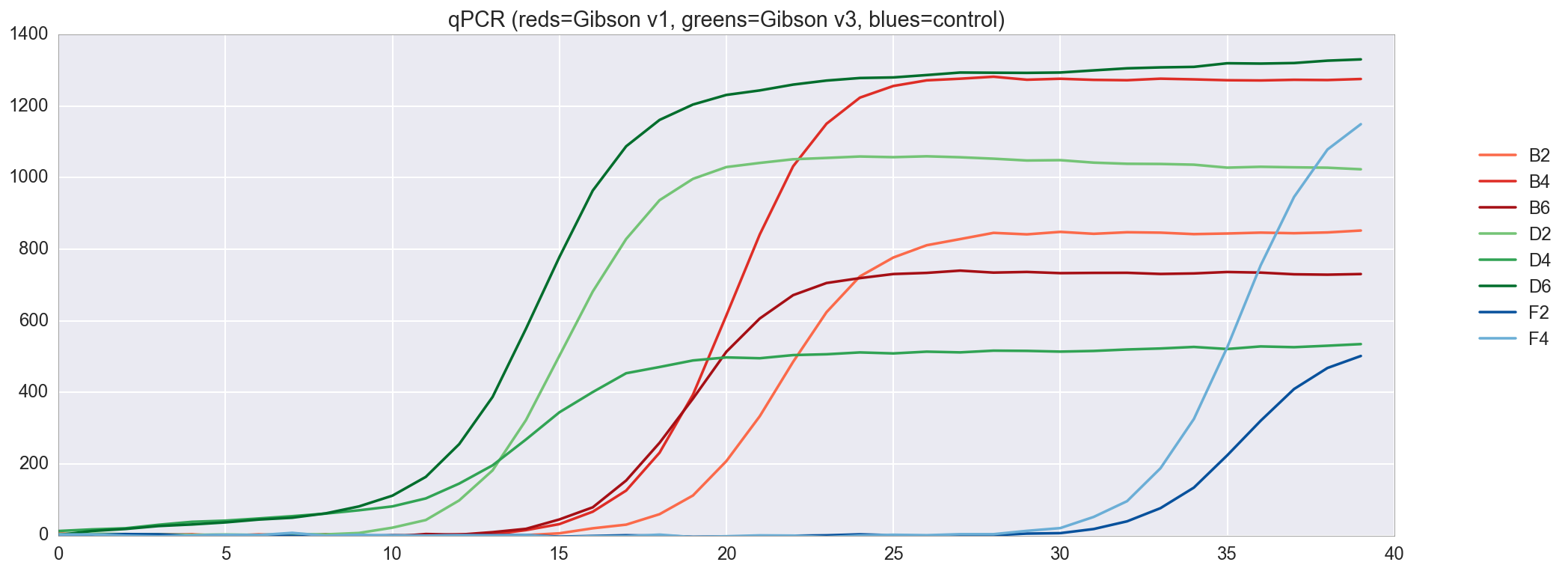 بشكل عام ، تبدو نتائج qPCR رائعة ، مع تضخيم جيد لكلا الإصدارين من مجموعة Gibson الخاصة بي وبدون تضخيم حقيقي في مجموعة التحكم. نظرًا لأن التجميع v3 أظهر نتيجة أفضل قليلاً من v1 ، فسنستخدمها الآن.
بشكل عام ، تبدو نتائج qPCR رائعة ، مع تضخيم جيد لكلا الإصدارين من مجموعة Gibson الخاصة بي وبدون تضخيم حقيقي في مجموعة التحكم. نظرًا لأن التجميع v3 أظهر نتيجة أفضل قليلاً من v1 ، فسنستخدمها الآن.النتائج: الجمعية جيبسون في هلام
يكون الجل أيضًا نظيفًا جدًا ، حيث يظهر نطاقات قوية أقل من 1 كيلو بايت في النطاقات B2 و B4 و B6 و D2 و D4 و D6: هذا هو الحجم الذي نتوقعه (يبلغ معدل الإدخال حوالي 740 نقطة أساس ، بينما تكون إشعال M13 حوالي 40 نقطة أساس للأعلى وللأسفل). الخط الثاني يتوافق مع الاشعال. يمكنك أن تكون متأكداً من ذلك ، لأن نطاقي F2 و F4 يحتويان فقط على الحمض النووي التمهيدي.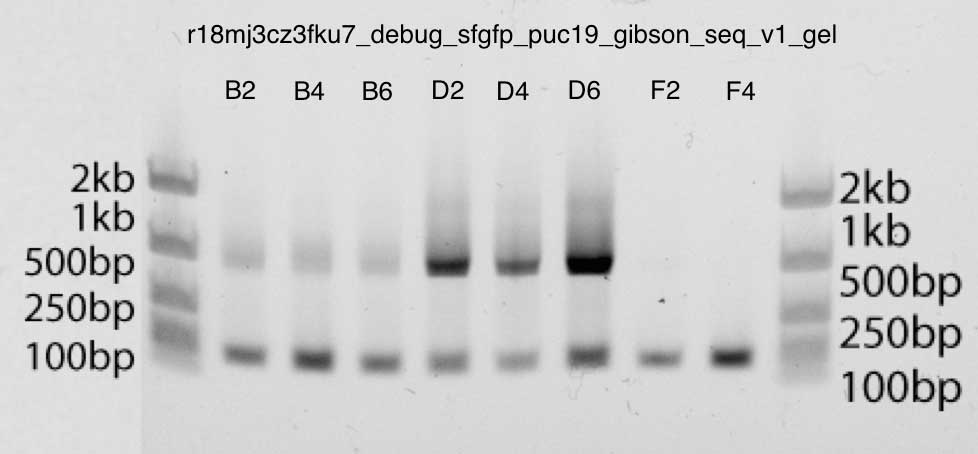 رحلان بولي أكريلاميد الكهربائي: يُظهر c3 Gibson assembly فرقًا أقوى (D2 ، D4 ، D6) ، وفقًا لبيانات qPCR أعلاه
رحلان بولي أكريلاميد الكهربائي: يُظهر c3 Gibson assembly فرقًا أقوى (D2 ، D4 ، D6) ، وفقًا لبيانات qPCR أعلاهالخطوة 4. التحول
التحول هو عملية تغيير الجسم عن طريق إضافة الحمض النووي. في هذه التجربة ، قمنا بتحويل E. coli باستخدام البلازميد pUC19 المعبر عن sfGFP.استخدام سهلة الاستخدام سلالة Zymo DH5α ميكس والعودة و بروتوكول الموصى بها . هذه السلالة هي جزء من المخزون النسخي القياسي. بشكل عام ، يمكن أن تكون التحويلات معقدة لأن الخلايا المختصة هشة إلى حد ما ، وبالتالي كلما كان البروتوكول أكثر بساطة وموثوقية ، كان ذلك أفضل. في مختبرات البيولوجيا الجزيئية العادية ، قد تكون هذه الخلايا المختصة مكلفة للغاية للاستخدام العام. Zymo ميكس وانطلق الخلايا مع بروتوكول بسيط
Zymo ميكس وانطلق الخلايا مع بروتوكول بسيطمشكلة في الروبوتات
هذا البروتوكول هو مثال جيد على مدى صعوبة تكييف البروتوكولات البشرية لاستخدامها بواسطة الروبوتات وكيف يمكن أن تفشل بشكل غير متوقع. تكون البروتوكولات في بعض الأحيان غامضة بشكل مدهش ("تأرجح الأنبوب من جانب إلى آخر") استنادًا إلى السياق العام لعلماء البيولوجيا الجزيئية ، أو قد تطلب فجأة معالجة متقدمة للصور ("تأكد من خلط الحبيبات"). لا يمانع الناس مثل هذه المهام ، لكن الروبوتات تحتاج إلى تعليمات أوضح.أظهر هذا التحول قضايا توقيت مثيرة للاهتمام. ينصح بروتوكول التحول بعدم بقاء الخلايا في درجة حرارة الغرفة لأكثر من بضع ثوانٍ ، ويجب تسخين اللوحة التي تحتوي على أنابيب إلى 37 درجة مئوية. من الناحية النظرية ، ترغب في بدء التسخين المسبق بحيث ينتهي بالتزامن مع هذا التحول ، لكن من غير الواضح كيف ستتعامل الروبوتات Transcriptic مع هذا الموقف - حسب علمي ، لا توجد طريقة لمزامنة خطوات البروتوكول بدقة. يبدو أن عدم وجود تحكم دقيق في الوقت يمثل مشكلة شائعة في البروتوكولات الآلية نظرًا لعدم المرونة النسبية للذراع الآلية وتعارضات الجدولة وما إلى ذلك. سيتعين علينا ضبط البروتوكولات وفقًا لذلك.عادة ما تكون هناك حلول معقولة: في بعض الأحيان تحتاج فقط إلى استخدام الكواشف المختلفة (على سبيل المثال ، المزيد من الخلايا المتشددة ، مثل Mix & Go أعلاه) ؛ في بعض الأحيان تقوم فقط برهن إجراءات الرهن العقاري بهامش (على سبيل المثال ، هز عشر مرات بدلاً من ثلاث) ؛ في بعض الأحيان تحتاج إلى التوصل إلى حيل خاصة للروبوتات (على سبيل المثال ، استخدم جهاز PCR لضربة الحرارة).بطبيعة الحال ، فإن الميزة الكبيرة هي أنه بمجرد أن يعمل البروتوكول مرة واحدة ، يمكنك عمومًا الاعتماد عليه مرارًا وتكرارًا. يمكنك حتى تحديد مدى موثوقية البروتوكول وتحسينه بمرور الوقت!اختبار التحول
قبل البدء في التحول باستخدام البلازميد المجمّع بالكامل ، أجري تجربة بسيطة للتأكد من أن التحويل باستخدام pUC19 المعتاد (أي بدون تجميع Gibson ودون إدخال DNA sfGFP) يعمل. يحتوي pUC19 على جين مقاومة الأمبيسيلين ، لذا فإن التحول الناجح يجب أن يسمح للبكتيريا بالنمو على ألواح تحتوي على هذه المضادات الحيوية.أقوم بنقل البكتيريا مباشرة إلى الجهاز اللوحي ("6-flat" في المصطلحات Transcriptic) ، حيث يوجد أمبيسيلين أم لا. أتوقع أن تحتوي البكتيريا المحولة على جين مقاومة الأمبيسلين وبالتالي ستنمو. يجب ألا تنمو البكتيريا غير المحولة.قانون """Simple transformation protocol: transformation with unaltered pUC19""" p = Protocol() experiment_name = "debug_sfgfp_puc19_gibson_v1" inv = { "water" : "rs17gmh5wafm5p",
✓ تحليل البروتوكول
43 تعليمات
3 حاويات
45.43 دولار
النتائج: اختبار التحول
في الصور التالية ، نرى أنه بدون وجود مضاد حيوي (صفيحة على اليسار) ، لوحظ نمو على جميع اللوحات الستة ، وإن كان ذلك بدرجات متفاوتة ، مما يسبب القلق. يبدو أن الروبوتات Transcriptic لا تتعامل حقًا مع التوزيع الموحد ، الأمر الذي يتطلب بعض البراعة.في ظل وجود المضادات الحيوية (الصفيحة اليمنى) ، هناك أيضًا نمو ، على الرغم من أنه غير متناسق مرة أخرى. تبدو الصفيحتان الأوليان اللتان تحتويان على مضادات حيوية غريبة ، مع نمو كبير ، وهو على الأرجح نتيجة لإضافة 55 ميكرولتر إلى هذه اللوحات مقارنة بـ 10 ميكرولتر على الأطباق بدون مضادات حيوية. هناك العديد من المستعمرات على اللوحة الثالثة ، وهذا هو ما كنت أتوقعه في جوهره على جميع اللوحات. يجب أن يكون هناك بعض النمو على اللوحات الثلاث الأخيرة ، لكن هذا لا يحدث. توضيحي الوحيد لهذه النتائج الغريبة هو أنني لم أخلط الخلايا والوسيلة بالقدر الكافي ، لذلك دخلت جميع الخلايا تقريبًا في الصفيحتين الأوليين.(لا يزال يتعين عليّ التحكم بشكل إيجابي في الأمبيسلين بواسطة بكتيريا غير متغيرة ، لكنني فعلت هذا بالفعل في تجربة سابقة ، لذلك أعرف أن مخزون الأمبيسيلين يجب أن يقتل هذه السلالةالقولونية E. . النمو أضعف بكثير على صفائح الأمبيسيلين ، على الرغم من وجود المزيد من البكتيريا ، كما هو متوقع).وعموما ، كان التحول يعمل بشكل جيد بما فيه الكفاية للاستمرار ، على الرغم من وجود بعض العيوب. تتحول صفائح من الخلايا باستخدام pUC19 بعد 18 ساعة: بدون مضاد حيوي (يسار) ومع مضاد حيوي (يمين)
تتحول صفائح من الخلايا باستخدام pUC19 بعد 18 ساعة: بدون مضاد حيوي (يسار) ومع مضاد حيوي (يمين)تحول المنتج بعد التجميع
نظرًا لأن مجموعة Gibson وتحول pUC19 البسيط يبدو أنهما يعملان ، يمكنك الآن تجربة التحول باستخدام بلازميد مركب بالكامل يعبر عن sfGFP.بالإضافة إلى الإدخال الذي تم جمعه ، سأضيف أيضًا القليل من IPTG و X-gal إلى اللوحات لرؤية التحويل الناجح باستخدام طريقة التحديد البيضاء والزرقاء . هذه المعلومات الإضافية مفيدة ، لأنه إذا حدث التحول باستخدام pUC19 المعتاد ، والذي لا يحتوي على sfGFP ، فسوف يستمر في إعطاء مقاومة للمضادات الحيوية.امتصاص ومضان
وفقًا لهذا الجدول ، يتوهج sfGFP بشكل أفضل عند أطوال موجية مثيرة تبلغ 485 نانومتر / 510 نانومتر. لقد وجدت أنه في Transcriptic ، 485/535 يعمل بشكل أفضل. أعتقد أن 485 و 510 متشابهان للغاية. أقيس نمو البكتيريا في 600 نانومتر ( OD600 ).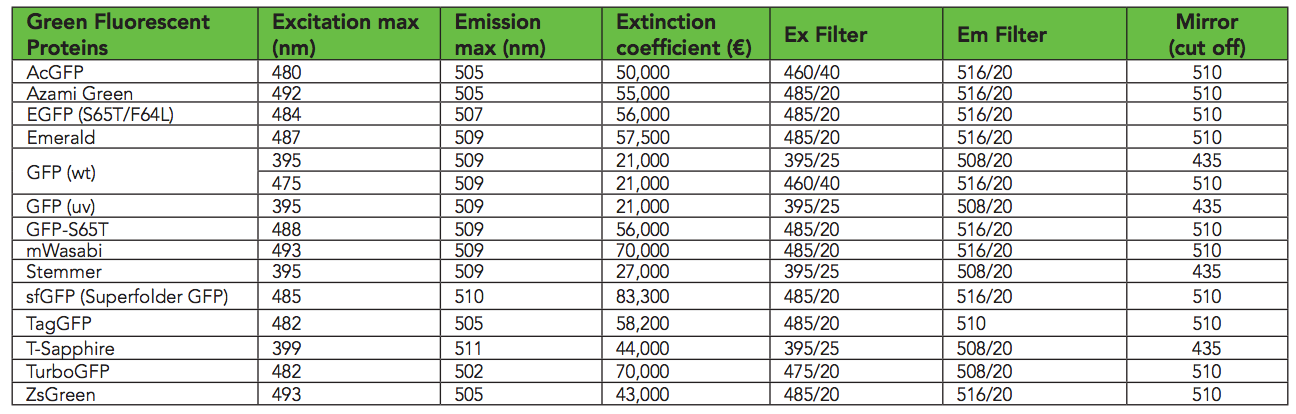 مجموعة متنوعة من GFP ( biotek )
مجموعة متنوعة من GFP ( biotek )IPTG و X-gal
يبلغ تركيز IPTG الخاص بي 1 مليونًا ويجب تخفيفه بنسبة 1: 1000. بدوره ، يجب أيضًا تخفيف X-gal بتركيز 20 ملغ / مل 1: 1000 (20 ملغ / ميكرولتر). لذلك ، في 2000µl رطل أضيف 2 ميكرولتر لكل منهما.وفقًا لبروتوكول واحد ، يجب أن تأخذ أولاً 40 ميكرولتر من X-gal بتركيز 20 ملغ / مل و 40 ميكرولتر من تركيز IPTG من 0.1 مم (أو 4 ميكرولتر من IPTG لكل 1M) ، واحتضانها لمدة 30 دقيقة. لم ينجح هذا الإجراء بالنسبة لي ، لذلك قمت فقط بخلط IPTG و X-gal والخلايا المقابلة واستخدمت هذا الخليط مباشرة.قانون """Full Gibson assembly and transformation protocol for sfGFP and pUC19 v1: Spread IPTG and X-gal onto plates, then spread cells v2: Mix IPTG, X-gal and cells; spread the mixture v3: exclude X-gal so I can do colony picking better v4: repeat v3 to try other excitation/emission wavelengths""" p = Protocol() options = { "gibson" : False,
المخزون: IPTG / IPTG / IPTG / IPTG / IPTG / IPTG 832.0: ميكروليتر {}
المخزون: sfgfp_puc19_gibson_v3_clone / sfgfp_puc19_gibson_v3_clone / sfgfp_puc19_gibson_v3_clone / sfgfp_puc19_gibson_v3_clone / sfgfp_puc19_gib}
✓ تحليل البروتوكول
40 تعليمات
8 حاويات
التكلفة الإجمالية: 53.20 دولار
وقت العمل: 17.35 دولار
الكواشف والمواد الاستهلاكية: 35.86 دولار جمع مستعمرة
عندما تنمو المستعمرات على صفيحة أمبيسيلين ، يمكنني "جمع" المستعمرات الفردية وزرعها على طبق من 96 أنبوبًا. هناك فريق خاص ( autopick ) لهذا في البروتوكول التلقائي .قانون """Pick colonies from plates and grow in amp media and check for fluorescence. v2: try again with a new plate (no blue colonies) v3: repeat with different emission and excitation wavelengths""" p = Protocol() options = {} for k, v in list(options.items()): if v is False: del options[k] experiment_name = "sfgfp_puc19_gibson_pick_v3" def plate_expid(val): """refer to the previous plating experiment's outputs""" plate_exp = "sfgfp_puc19_gibson_plates_v4" return "{}_{}".format(plate_exp, val)
✓ تحليل البروتوكول
62 تعليمات
8 حاويات
التكلفة الإجمالية: 66.38 دولار
وقت العقد: 57.59 دولار
الكواشف والمواد الاستهلاكية: 8.78 دولار
النتائج: مستعمرة الحصاد
أظهرت الشاشة ذات اللون الأزرق والأبيض تمامًا ، بشكل أساسي ، مستعمرات بيضاء على ألواح بها مضاد حيوي (1-4) وأزرق فقط على ألواح بدون مضاد حيوي (5-6). هذا بالضبط ما كنت أتوقعه ، وكنت سعيدًا برؤيته ، خاصة وأنني استخدمت IPTG و X-gal ، التي أرسلتها إلى Transcriptic.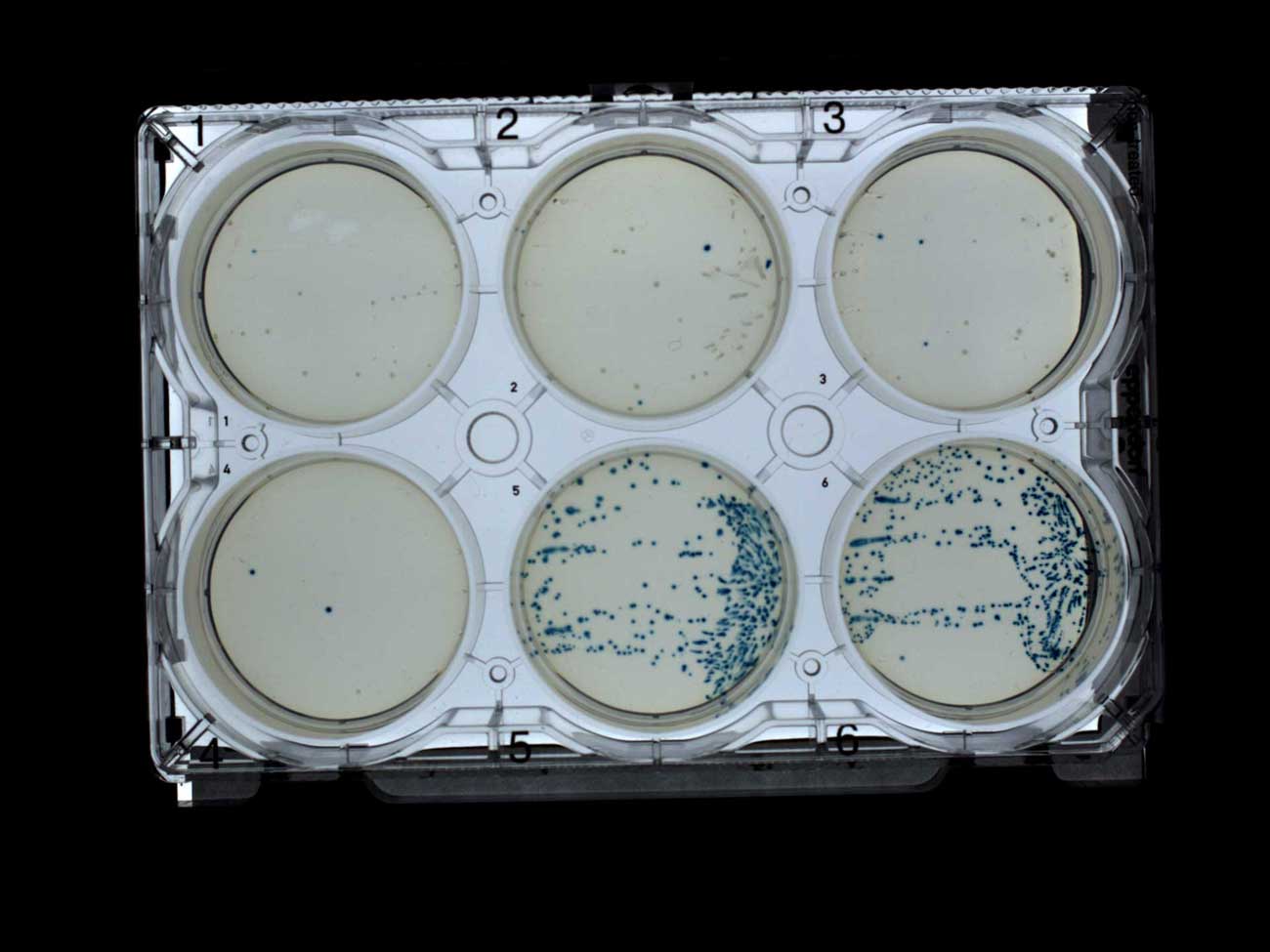 الفحص عن طريق اختيار لوحات بيضاء زرقاء مع أمبيسيلين (1-4) وبدون مضاد حيوي (5-6)ومع ذلك ، فإن روبوت تجميع المستعمرات لم ينجح بشكل جيد مع هذه المستعمرات ذات اللون الأزرق الأبيض. تم إنشاء الصورة أدناه من خلال طرح صور متتالية من اللوحات بعد كل جولة من تحديد اللوحة وزيادة التباين في الاختلافات (في GraphicsMagick ). وبهذه الطريقة ، يمكنني تصور المستعمرات التي تم جمعها (على الرغم من أنها ليست مثالية ، نظرًا لأن المستعمرات التي تم جمعها لم تتم إزالتها بالكامل).لقد وقعت الصورة أيضًا بعدد المستعمرات التي جمعها روبوت النسخ. كان من المفترض أنه سيجمع 10 مستعمرات كحد أقصى من اللوحات الخمسة الأولى. ومع ذلك ، بشكل عام ، تم جمع العديد من المستعمرات ، وهذه عادة ما تكون مستعمرات زرقاء. تمكن الروبوت فقط من العثور على عشرة مستعمرات على لوحة التحكم مع المستعمرات الزرقاء فقط. نظريتي العملية هي أن روبوت جمع مستعمرة يجمع المستعمرات الزرقاء بشكل مفضل لأنها أكثر تباينًا.
الفحص عن طريق اختيار لوحات بيضاء زرقاء مع أمبيسيلين (1-4) وبدون مضاد حيوي (5-6)ومع ذلك ، فإن روبوت تجميع المستعمرات لم ينجح بشكل جيد مع هذه المستعمرات ذات اللون الأزرق الأبيض. تم إنشاء الصورة أدناه من خلال طرح صور متتالية من اللوحات بعد كل جولة من تحديد اللوحة وزيادة التباين في الاختلافات (في GraphicsMagick ). وبهذه الطريقة ، يمكنني تصور المستعمرات التي تم جمعها (على الرغم من أنها ليست مثالية ، نظرًا لأن المستعمرات التي تم جمعها لم تتم إزالتها بالكامل).لقد وقعت الصورة أيضًا بعدد المستعمرات التي جمعها روبوت النسخ. كان من المفترض أنه سيجمع 10 مستعمرات كحد أقصى من اللوحات الخمسة الأولى. ومع ذلك ، بشكل عام ، تم جمع العديد من المستعمرات ، وهذه عادة ما تكون مستعمرات زرقاء. تمكن الروبوت فقط من العثور على عشرة مستعمرات على لوحة التحكم مع المستعمرات الزرقاء فقط. نظريتي العملية هي أن روبوت جمع مستعمرة يجمع المستعمرات الزرقاء بشكل مفضل لأنها أكثر تباينًا. ألواح الفحص للاختيار بين الأزرق والأبيض مع الأمبيسلين (1-4) وبدون مضاد حيوي (5-6) ، مما يشير إلى عدد المستعمرات المجمعةخدم الفحص الأزرق والأبيض لغرض معين. أظهر أن معظم المستعمرات تتحول بشكل صحيح. على الأقل هناك الإدراج. ومع ذلك ، من أجل جمع أفضل مستعمرة ، كررت التجربة بدون X-gal.فقط مع المستعمرات البيضاء نجح جامع الروبوت في تجميع عشر مستعمرات بنجاح من كل من الألواح الخمسة الأولى. يمكن افتراض أنه في معظم المستعمرات التي تم جمعها هناك عمليات إدخال ناجحة.
ألواح الفحص للاختيار بين الأزرق والأبيض مع الأمبيسلين (1-4) وبدون مضاد حيوي (5-6) ، مما يشير إلى عدد المستعمرات المجمعةخدم الفحص الأزرق والأبيض لغرض معين. أظهر أن معظم المستعمرات تتحول بشكل صحيح. على الأقل هناك الإدراج. ومع ذلك ، من أجل جمع أفضل مستعمرة ، كررت التجربة بدون X-gal.فقط مع المستعمرات البيضاء نجح جامع الروبوت في تجميع عشر مستعمرات بنجاح من كل من الألواح الخمسة الأولى. يمكن افتراض أنه في معظم المستعمرات التي تم جمعها هناك عمليات إدخال ناجحة. مستعمرات تنمو على ألواح مع الأمبيسلين (1-4) وبدون مضاد حيوي (5-6)
مستعمرات تنمو على ألواح مع الأمبيسلين (1-4) وبدون مضاد حيوي (5-6)النتائج: التحول مع المنتج تجميعها
بعد زراعة 50 مستعمرة مختارة على طبق من 96 أنبوبًا لمدة 20 ساعة ، أقيس التألق للتحقق من تعبير sfGFP. يستخدم Transcriptic قارئ Tecan Infinite لقياس التألق والامتصاص (والإشراق ، إذا أردت) .من الناحية النظرية ، في أي مستعمرة ، يجب تجميع البلازميد مع النمو ، لأنه يحتاج إلى مقاومة للمضادات الحيوية ، ويعبر كل بلازميد يتم جمعه عن sfGFP. هناك بالفعل العديد من الأسباب وراء عدم حدوث ذلك ، لأسباب ليس أقلها أنه يمكنك فقد الجين sfGFP من البلازميد دون فقد مقاومة الأمبيسيلين. للبكتيريا التي تفقد جين sfGFP ميزة في الاختيار على منافسيها ، لأنها لا تنفق طاقة إضافية ، ولكن مع الأخذ في الاعتبار عدد كافٍ من أجيال النمو ، فإن هذا سيحدث بالتأكيد.أقوم بجمع بيانات الامتصاص (OD600) ومضان كل أربع ساعات لمدة 20 ساعة (حوالي 60 جيلًا). for t in [0,4,8,12,16,20]: abs_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_abs_{}.csv".format(t), index_col="Well") flr_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_fl2_{}.csv".format(t), index_col="Well") if t == 0: new_data = abs_data.join(flr_data) else: new_data = new_data.join(abs_data, rsuffix='_{}'.format(t)) new_data = new_data.join(flr_data, rsuffix='_{}'.format(t)) new_data.columns = ["OD 600:nanometer_0", "Fluorescence_0"] + list(new_data.columns[2:])
نضع على الرسم البياني بيانات الساعة العشرين وآثار القياسات السابقة. في الواقع ، أنا مهتم فقط بأحدث البيانات ، لأنه عندئذ يجب ملاحظة ذروة مضان. svg = [] W, H = 800, 500 min_x, max_x = 0, 0.8 min_y, max_y = 0, 50000 def _toxy(x, y): return W*(x-min_x)/(max_x-min_x), HH*(y-min_y)/(max_y-min_y) def _topt(x, y): return ','.join(map(str,_toxy(x,y))) ab_fls = [[row[0]] + [list(row[1])] for row in new_data.iterrows()]
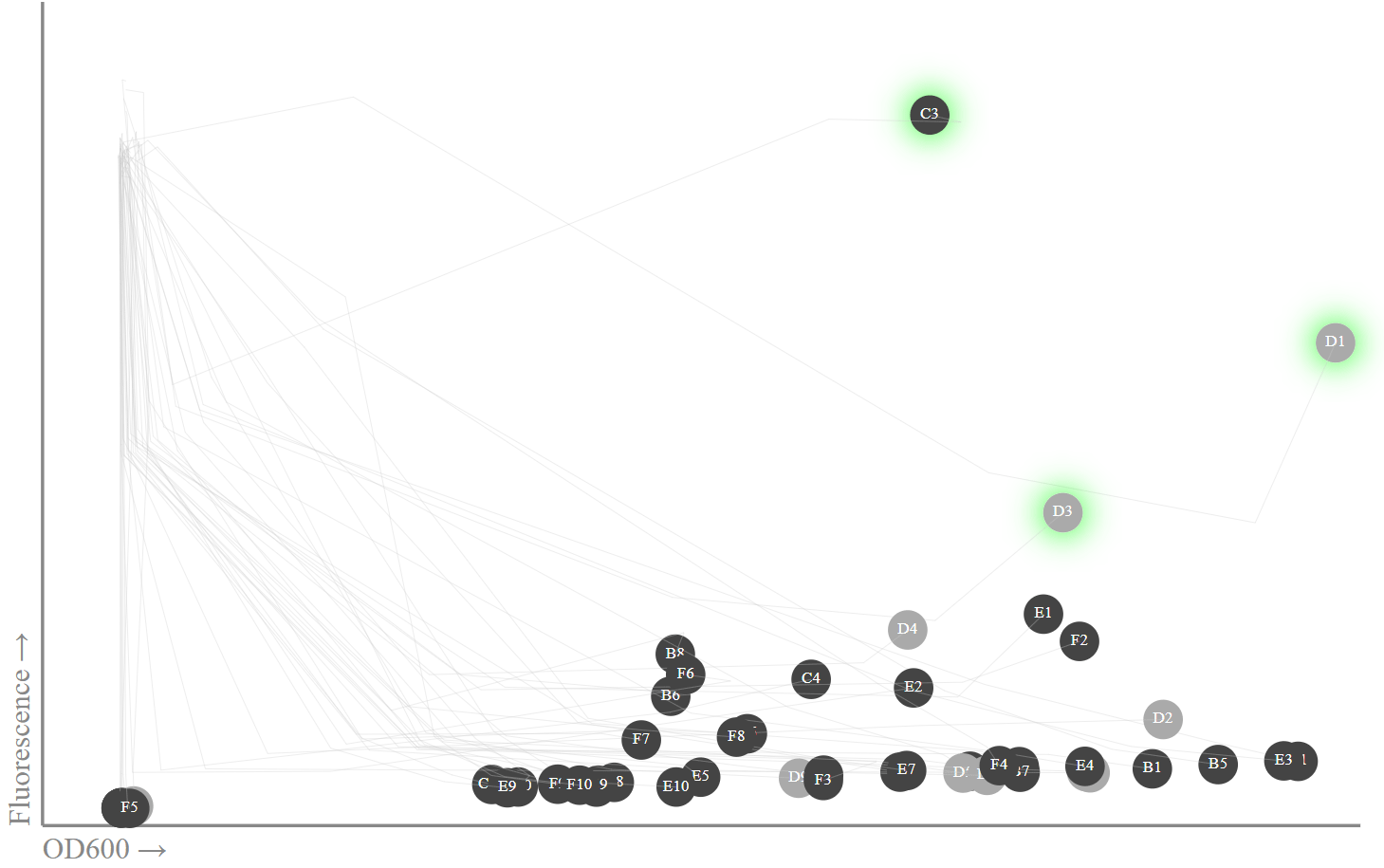 الإسفار و OD600: المستعمرات مع الأمبيسلين سوداء ، ومستعمرات السيطرة بدون الأمبيسلين رمادية. يتم تمييز المستعمرات باللون الأخضر ، حيث أكدت أن تسلسل بروتين sfGFP صحيح ، فقمبتشغيل miniprep لاستخراج DNA البلازميد ، ثم تسلسل Sanger باستخدام الاشعال M13. لسوء الحظ ، لسبب ما ، يتوفر miniprep حاليًا فقط من خلال بروتوكول الويب Transcriptic ، وليس عبر بروتوكول تلقائي. سأقوم بتسلسل الأنابيب الثلاثة مع أعلى قراءات مضان (C1 ، D1 ، D3) والثلاثة الأخرى (B1 ، B3 ، E1) ، محاذاة التسلسلات مع sfGFP باستخدام العضلات .في الأنابيب C1 و D3 و D3 ، فإن المطابقة المثالية لتسلسل sfGFP الأصلي الخاص بي ، بينما في B1 و B3 و E1 ، تفشل الطفرات الخشنة أو المحاذاة ببساطة.
الإسفار و OD600: المستعمرات مع الأمبيسلين سوداء ، ومستعمرات السيطرة بدون الأمبيسلين رمادية. يتم تمييز المستعمرات باللون الأخضر ، حيث أكدت أن تسلسل بروتين sfGFP صحيح ، فقمبتشغيل miniprep لاستخراج DNA البلازميد ، ثم تسلسل Sanger باستخدام الاشعال M13. لسوء الحظ ، لسبب ما ، يتوفر miniprep حاليًا فقط من خلال بروتوكول الويب Transcriptic ، وليس عبر بروتوكول تلقائي. سأقوم بتسلسل الأنابيب الثلاثة مع أعلى قراءات مضان (C1 ، D1 ، D3) والثلاثة الأخرى (B1 ، B3 ، E1) ، محاذاة التسلسلات مع sfGFP باستخدام العضلات .في الأنابيب C1 و D3 و D3 ، فإن المطابقة المثالية لتسلسل sfGFP الأصلي الخاص بي ، بينما في B1 و B3 و E1 ، تفشل الطفرات الخشنة أو المحاذاة ببساطة.ثلاث مستعمرات مضيئة
النتائج جيدة ، على الرغم من أن بعض الجوانب مفاجئة. على سبيل المثال ، يبدأ قارئ التألق ، دون سبب واضح ، في وقت 0 بقيم عالية جدًا (40000 قطعة). بحلول الساعة العشرين ، كان قد هدأ إلى نمط أكثر منطقية مع وجود علاقة واضحة بين OD600 ومضان (أفترض بسبب وجود تداخل بسيط في الأطياف) ، بالإضافة إلى بعض الانبعاثات مع مضان عالية. من المثير للدهشة ، أن تكون واحدة أو ثلاثة أو ربما 11-15 انبعاثات.توجد بعض الأنابيب ذات قيم التألق العالية في أنابيب التحكم (أي بدون الأمبيسلين ، فهي رمادية اللون) ، وهو أمر يثير الدهشة نظرًا لعدم وجود ضغط اختيار في هذه الأنابيب ، وبالتالي ، يمكن توقع فقد البلازميد).استنادًا إلى بيانات التألق ونتائج التسلسل ، يبدو أن ثلاثة فقط من 50 مستعمرة تنتج sfGFP و fluoresce. هذا ليس بقدر ما كنت أتوقع. ومع ذلك ، نظرًا لوجود ثلاث مراحل نمو منفصلة (على اللوح ، في المختبر ، من أجل miniprep) ، فقد خضع حوالي 200 جيل من النمو لهذه المرحلة من الخلية ، لذلك كان هناك الكثير من الفرص لحدوث طفرات.يجب أن تكون هناك طرق لجعل العملية أكثر كفاءة ، خاصة وأنني بعيد عن خبير في هذه البروتوكولات. ومع ذلك ، فقد أنتجنا بنجاح خلايا محولة مع تعبير عن GFP الهندسي باستخدام فقط رمز Python!الجزء الثالث: الاستنتاجات
السعر
بناءً على كيفية القياس ، كانت تكلفة هذه التجربة حوالي 360 دولارًا ، وليس بما في ذلك أموال تصحيح الأخطاء:- 70 دولارا لتوليف الحمض النووي
- $32 PCR
- $31
- $32
- $53
- $67
- $75 3 miniprep'
أعتقد أنه يمكن تخفيض التكلفة إلى 250-300 دولار مع بعض التحسينات. على سبيل المثال ، المجموعة الآلية المكونة من 50 مستعمرة مكلفة بشكل مثير للريبة وربما يمكن التخلي عنها.في تجربتي ، يبدو هذا السعر مرتفعًا بالنسبة لبعض (علماء الأحياء الجزيئية) ومنخفضًا بالنسبة للآخرين (أشخاص تكنولوجيا المعلومات). بما أن Transcriptic يشحن فقط كواشف قائمة الأسعار ، فإن الفرق الرئيسي في التكاليف هو العمالة. الروبوت بالفعل رخيص للغاية في ساعة واحدة ، وقال انه لا يمانع في الاستيقاظ في منتصف الليل لتصوير لوحة. بمجرد الموافقة على البروتوكولات ، يكون من الصعب تخيل أنه حتى طالب الدراسات العليا سيكون أرخص ، لا سيما بالنظر إلى تكاليف الفرصة البديلة.من أجل الوضوح ، أنا أتحدث فقط عن استبدال البروتوكولات الروتينية. بطبيعة الحال ، سيستمر علماء الأحياء الجزيئيون المؤهلون في تطوير البروتوكولات المتقدمة ، لكن العديد من مجالات العلوم المثيرة تستخدم بروتوكولات روتينية مملة. حتى وقت قريب ، أنتجت العديد من المختبرات قليل النوكليوتيدات الخاصة بها ، ولكن الآن قليل من الناس قلقون بشأن هذا: إنه لا يكلف أي شخص في أي وقت ، ولا حتى طلاب الدراسات العليا ، عندما يرسلهم IDT إليكم في غضون بضعة أيام.مختبرات الروبوتية: إيجابيات وسلبيات
من الواضح ، أنا أؤمن حقًا بمستقبل المختبرات الآلية. هناك بعض الأشياء المضحكة والمفيدة حقًا في تجربة الروبوتات ، خاصة إذا كنت مشتركًا في الحوسبة ولديك حساسية من قفازات اللاتكس والعمل اليدوي:- ! , . autoprotocol, .
- . 100 , .
- , , PCR. , , ? / ? , , , « 2-3 ». ?
- . . , .
- . .
- التعبير . يمكنك استخدام بناء جملة البرمجة لترميز الخطوات المكررة أو منطق المتفرعة. على سبيل المثال ، إذا كنت ترغب في الاستغناء عن 1 إلى 96 ميكرولتر من الكاشف و (96 - x) ميكرولتر من الماء في لوحة مكونة من 96 أنبوبًا ، يمكن كتابة هذا باختصار.
- بيانات آلة قابلة للقراءة . يتم دائمًا إرجاع البيانات ذات النتائج بتنسيق csv أو بتنسيق آخر مناسب لمعالجة الآلة.
- التجريد . من الناحية المثالية ، يمكنك تشغيل البروتوكول بالكامل بغض النظر عن الكواشف المستخدمة أو أسلوب الاستنساخ واستبدال شيء ما إذا لزم الأمر ، إذا كان يعمل بشكل أفضل.
بالطبع ، هناك بعض العيوب ، خاصة وأن الأدوات بدأت للتو في التطور. إذا قارنا الإنترنت ، فنحن في منطقة 1994:- Transcriptic — . , , , . , , .
- — Transcriptic.
- , . Transcriptic ( , , ).
- بالنسبة للعديد من المختبرات ، قد يكون استخدام معمل سحابي أغلى من مجرد أخذ طالب دراسات عليا للقيام بهذا العمل (التكلفة الحدية للساعة: ~ $ 0). يعتمد ذلك على ما إذا كان المختبر بحاجة إلى أيدي طالب الدراسات العليا أو ذكائه.
- لم يتم تجربة النسخة التجريبية في نهاية الأسبوع. يمكنك فهمهم ، لكن هذا قد يكون غير مريح ، حتى لو كان لديك مشروع صغير.
برنامج لصنع البروتينات
على الرغم من وجود الكثير من التعليمات البرمجية والكثير من عمليات تصحيح الأخطاء ، أعتقد أنه من الممكن إنشاء نوع من البرامج التي تأخذ سلسلة من البروتينات كمدخلات وتخلق بكتيريا مع التعبير عن هذا البروتين في الإخراج.لكي ينجح هذا ، يجب أن تحدث عدة أشياء:- التكامل الحقيقي بين Twist / IDT / Gen9 مع Transcriptic (ربما سيكون بطيئًا بسبب انخفاض الطلب الحالي).
- , , , , . .
- ( NEB, IDT) (, primer3 ).
في العديد من التطبيقات ، تريد أيضًا تنقية البروتين (عبر عمود ) ، أو ربما تجعل البكتيريا تفرزه . لنفترض أنه يمكننا القيام بذلك قريبًا في معمل سحابي ، أو أنه يمكننا إجراء تجارب حية (أي داخل خلية بكتيرية).هناك العديد من الاحتمالات لكي يعمل البروتوكول فعليًا بشكل أفضل من البشر ، على سبيل المثال: تصميم المروجين و RBS لتحسين التعبير المخصص لتسلسلك ؛ إحصائيات عن احتمال نجاح تجربة تستند إلى تجارب قابلة للمقارنة ؛ تحليل الجل الآلي.لماذا كل هذا؟
بعد كل هذا ، قد لا يكون من الواضح تمامًا سبب إنشاء هذا البروتين. إليك بعض الأفكار:- - //, .
- , , .
- in vivo split-GFP .
- scFv . scFvs - .
- BiTE , ( , ).
- اصنع لقاحًا محليًا يدخل الجسم من خلال بصيلات الشعر (لا أوصي بتجربة ذلك في المنزل).
- قم بتطفير البروتين بمئات الطرق المختلفة وشاهد ما يحدث. ثم النطاق إلى 1000 أو 10000 طفرات؟ ربما تميز طفرات GFP؟
للحصول على أفكار جديدة حول ما هو ممكن مع تصميم البروتين ، انظر إلى مئات مشاريع iGEM .في النهاية ، أود أن أشكر Transcriptic Ben Miles لمساعدته في إكمال هذا المشروع.