في
المقالة المفصلة السابقة حول
الجينوم الكامل ، وعدنا بنشر ثلاث مشاكل وإجراء اختبار للشخص الذي حل المشكلات الثلاثة أولاً بشكل صحيح. في الوقت نفسه ، نقدم أمثلة على كيفية التعامل مع البيانات الجينية في هذه المهام. اليوم ننشر الأول.

في
المقالة الأولى
، تبادلنا المعلومات والروابط المفيدة المفيدة للعمل مع بيانات المعلوماتية الحيوية. نوصي بقراءته أولاً إذا فاتتك.
تنصليتم العمل مع البيانات الوراثية على أنظمة Unix (Linux ، macOS) ، حيث أن بعض الأوامر والبرامج غير متوفرة على Windows. لذلك ، لمستخدمي Windows ، أحد أبسط الحلول هو استئجار جهاز Linux افتراضي.
يتم تنفيذ جميع العمليات الموضحة أدناه في سطر الأوامر - المحطة الطرفية. قبل البدء ، تعرف على كيفية العمل في محطة تشغيل نظام التشغيل الخاص بك واستخدام الأوامر ، لأن بعضها يمكن أن يضر نظام التشغيل والبيانات الخاصة بك.
البرامج المطلوبة
لقد جمعنا
صورة جهاز افتراضي (VM) مع جميع البرامج اللازمة على Yandex.Cloud. سجل في Yandex.Cloud ، في حسابك في قسم Compute Cloud ، انقر فوق إنشاء VM. كصورة عامة ، حدد 1000 جينوم من كتالوج تحليل بيانات أطلس.
تكوين VM: 100 ٪ 2vCPU ، 8GB RAM ، 20GB HDD. عند إنشاء VM ، يجب عليك إدخال بيانات الدفع ، ولكن لا يتم شطب أي شيء من الحساب. بداية ومنحة إضافية على كلمة رمز كافية للعمل مع VM والصورة من أطلس حتى 31 ديسمبر 2019 مجانا. لتلقي منحة لإكمال المهام ، أرسل كلمة الكود "ATLAS" إلى
دعم Yandex.Cloud .
ملاحظة: المنحة صالحة لمستخدمي Yandex.Cloud الجدد الذين قاموا بالتسجيل منذ 18 ديسمبر 2019 أو لأولئك الذين ما زالوا لديهم فترة تجريبية ولديهم منحة بدء. كلمة الكود ATLAS صالحة مرة واحدة فقط.
قم أولاً بإنشاء مفتاح ssh على الكمبيوتر المحلي الذي تخطط منه للاتصال بـ VM:
ssh-keygen -o -t rsa -b 4096 -C "my-local-machine" -f ~/.ssh/yandex-cloud -a 100
لا تنسَ نسخ محتويات ملف
~/.ssh/yandex-cloud.pub إلى الإطار المناسب عند إنشاء VM.
إذا كنت ترغب في تثبيت البرنامج على جهاز الكمبيوتر الخاص بك ، ستجد أدناه جميع معلومات التثبيت. إذا قررت استخدام Yandex.Cloud ، فقم بإنشاء VM وانتقل إلى القسم التالي.
طقطقة
Plink عبارة عن حزمة برمجية لمعالجة البيانات الجينية والبحث عن ارتباط الجينوم الواسع (GWAS). تم تطويره من قبل عالم الوراثة شون بورسيل (شون بورسيل). منذ عام 2008 ، بمساعدة Plink ، تم إجراء المئات من GWAS في جميع أنحاء العالم ، وكانت نتائج أفضل ما يستخدمه Atlas كمصدر بيانات للخوارزميات لحساب مخاطر الأمراض.
يقدم Plink مجموعة من الأدوات لتخزين بيانات التنميط الجيني وتحويلها والبحث عنها. يسمح Plink أيضًا بمعالجة إحصائية وتحليل اختلال الارتباط (LD) ، وتحديد الهوية من خلال النسب (IBD) وتحليل الهوية (الحالة من خلال IBS) ، واختبارات التقسيم الطبقي للسكان ورسائل الربو - تفاعل العديد من الاختلافات الوراثية فيما بينهم.
يتم استخدام IBD و IBS لتحليل التركيبة السكانية وتحديد القرابة.
مثال على ذلك ، الاختلافات في rs7412 و rs429358 في جين APOE ، وهو مزيج معين من المتغيرات التي تزيد بشكل حاد من خطر الإصابة بمرض الزهايمر ، في حين أن كل متغير على حدة لا يقدم سوى مساهمة صغيرة في الخطر.
قم بتنزيل الإصدار الثابت من Plink من
الموقع الرسمي.
BCFtools
BCFtools هي مجموعة من الأدوات المساعدة لمعالجة البيانات الوراثية بتنسيق VCF ونظيرتها الثنائية BCF. تتضمن قائمة التطبيقات الممكنة لحزمة BCFtools التعليقات التوضيحية والترشيح ودمج وتقسيم ملفات VCF / BCF ، وإيجاد التقاطعات الخاصة بهم ، والفهرسة ، والبحث الانتقائي ، والفرز ، وحساب الإحصائيات ، إلخ.
للتثبيت ، قم بما يلي:
git clone git://github.com/samtools/htslib.git git clone git://github.com/samtools/bcftools.git cd bcftools
يتم وصف عملية التثبيت بمزيد من التفاصيل
هنا .
KING
تُستخدم حزمة KING (الاستدلال المستند إلى القرابة لل Gwas) في الدراسات السكانية عند العمل مع البيانات من البحث على نطاق الجينوم عن الجمعيات لتحديد علاقات القرابة في البيانات المدروسة. في هذه المهمة ، ستساعد KING في تحديد درجة قرابة عدة عينات من مشروع 1000 Genomes.
يمكنك تنزيله من
هنا . لحل المشكلات ، يتوفر دليل KING
هنا .
يتم وصف جميع الأخطاء التي قد تحدث أثناء العمل باستخدام الأدوات في Stackoverflow أو نظيرتها الأحيائية - Biostars .
البيانات المستخدمة
للحصول على إرشادات ، نستخدم البيانات المفتوحة
من مشروع 1000 Genomes. للتحليل ، اخترنا 10 عينات مع معلومات حول الأنماط الجينية من حوالي 85 مليون الاختلافات التي تم الحصول عليها عن طريق تحليل بيانات NGS محاذاة مع نسخة من الجينوم المرجعي GRCh37. وتظهر العلاقات الأسرية والسكان عينة في الشكل 1.
 الشكل 1
الشكل 1 نسب المستخدمة في عينات VCF. المربع يتوافق مع الجنس من الذكور ، الدائرة إلى الأنثى. الخط المنقط يعني صلة القرابة من الدرجة الثانية غير المحددة.
يحيط علما
يسمح لك تنسيق VCF بتخزين معلومات حول حقل شخص كرقم واحد ، إذا كانت هذه المعلومات معروفة أثناء إنشاء VCF. يبدو الأمر كما يلي: يحتوي حقل GT (النمط الوراثي ، النمط الوراثي) للسجلات من كروموسوم X على قيمة عددية واحدة تتوافق مع أليل واحد ، للرجال واثنان للنساء. إذا لم تكن هناك معلومات حول الحقل البيولوجي للعينة المتسلسلة ، فسيحتوي حقل GT افتراضيًا على قيمتين رقميتين (مظللة باللون الأحمر في الشكل 2).
في ملفات VCF المستخدمة في هذا الدليل ، يتم استبعاد كروموسوم Y ، لكن وجود كروموسوم Y في ملف VCF لا يعني دائمًا أن العينة المتسلسلة تحتوي عليه بالفعل. ويرجع ذلك إلى مناطق جسمية زائفة (PARs) ، وهي متطابقة لكروموسومات X و Y وتقع في نهايتها.
عادة لا تحتوي الكروموسومات المختلفة على مناطق متطابقة طويلة (متجانسة) ، ومع ذلك ، فإن كروموسومات X و Y تمتلك مثل هذه المناطق عدة ملايين من الأزواج الأساسية منذ البداية (PAR1) ونهاية (PAR2). لذلك ، عند تحليل بيانات NGS لدى الرجال في مناطق PAR ، يوجد أليلان (واحد لكل كروموسوم جنسي) ، وفي النساء ، قد تظهر الأنماط الوراثية في مناطق PAR من كروموسوم Y ، على الرغم من أنها في الواقع أنماط وراثية من كروموسوم X.
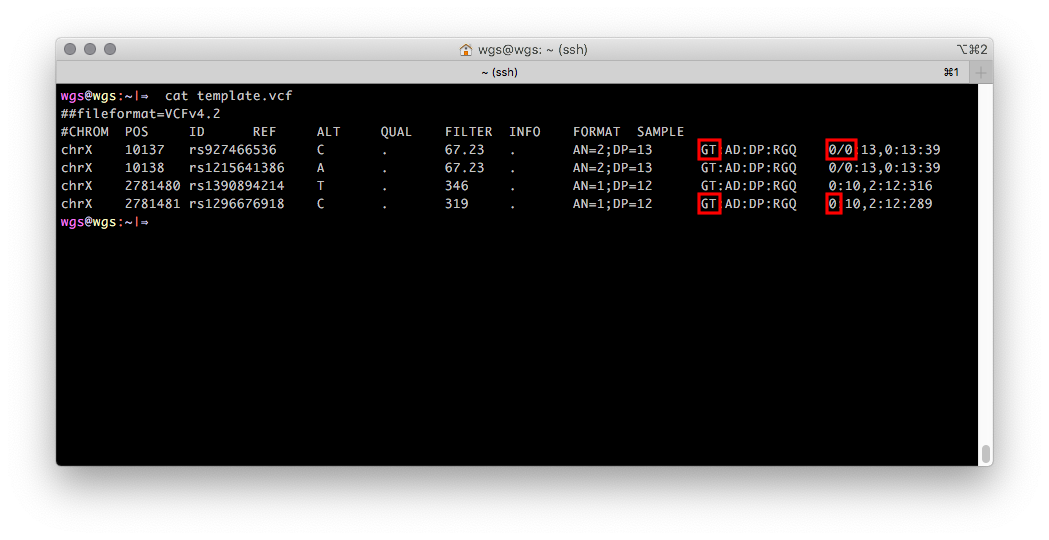 الشكل 2
الشكل 2 ملف VCF به أنماط وراثية من كروموسوم X لرجل من منطقة PAR1 (أول مدخلين) ومنطقة غير جسمية مزيفة (آخر إدخالين).
الوحدة التعليمية
الجنس الوراثي هو مجموعة من الكروموسومات الجنسية التي تتوافق مع مظاهر الخصائص الجنسية الأولية والثانوية من النوع الذكوري أو الأنثوي. عادة ، يكون لدى الرجال كروموسوم X واحد وكروموسوم Y ، في حين أن النساء لديهما كروموسومات X. مع اضطرابات مختلفة في تكوين الخلايا الجرثومية والبيض والحيوانات المنوية ، يمكن أن يولد طفل مع مجموعة ممتازة من الكروموسومات الجنسية للوالدين ، مما يؤدي في كثير من الأحيان إلى اضطرابات النمو الخصائص الجنسية الأولية والثانوية.
الشذوذان الجنسيان الأكثر شيوعًا هما: متلازمة تيرنر (مجموعة من الكروموسومات X0 ، أي كروموسوم X واحد فقط) ومتلازمة كلاينفلتر (مجموعة من الكروموسومات XXY).
الأليل هو واحد أو أكثر من النيوكليوتيدات الموجودة في أي موضع في الجينوم وله بديل. يستخدم المفهوم لوصف الأنماط الجينية. التمييز بين الأليلات المرجعية والبديلة. يتم تخزين كل منهم في ملف VCF في حقول REF و ALT ، على التوالي.
تحديد الجنس
لمستخدمي Yandex.Cloudيتم تخزين جميع البيانات لاستكمال المهام اليدوية والمستقلة على Yandex.Cloud باستخدام الهيكل الموضح أدناه. يحتوي مجلد
Tutorial على ملف VCF اللازم لإكمال الدليل ، وهو مجلد
Test للمهام المستقلة. يحتوي المجلد
Technical على ملفين مع قائمة
rsids_for_subsetting.txt الاختلافات الوراثية:
rsids_for_subsetting.txt استخدام
rsids_for_subsetting.txt في الدليل ، وقد تكون هناك حاجة إلى مهام للتنفيذ المستقل ،
external_interpretation_rsids.txt في المستقبل عند الحصول على التسلسل على نطاق الجينوم في الأطلس لتحميل بيانات التنميط الجيني إلى خدمات الطرف الثالث. يحتوي المجلد "
Tools ، من بين أشياء أخرى ، على اثنين من البرامج النصية المستخدمة في المهام 2 و 3.
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
سيتم إنشاء مجلد في الدليل
/home على Yandex.Cloud VM ، والذي يتوافق اسمه مع اسم المستخدم المحدد في مرحلة إنشاء VM. انسخ كل شيء من دليل
/home/ubuntu إلى الدليل الخاص بك من خلال الأوامر التالية:
cd ~ cp -r /home/ubuntu/* ./
لبقيةعند العمل على جهاز كمبيوتر شخصي ، يمكنك تنزيل الملفات الضرورية للمهمة الأولى من
الرابط . يدعم الأرشيف الذي تم تنزيله بنية تخزين ملفات مماثلة لتلك المستخدمة في Yandex.Cloud:
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
قم بفك
atlas_wgs_contest.tar.gz أرشيف
atlas_wgs_contest.tar.gz باستخدام الأمر
tar -xvzf atlas_wgs_contest.tar.gz تشغل ملفات VCF لأداء المهام في شكل غير مؤرخ حوالي 19 غيغابايت لكل منهما ، لذلك ، لتوفير مساحة ، نوصي بالعمل فقط مع الأرشيف. جميع البرامج المذكورة أعلاه قادرة بالفعل على العمل مع بيانات VCF المضغوطة. بالإضافة إلى ذلك ، لا تحتاج إلى القيام بأي شيء.
لتحديد جنس الموضوع ، تحتاج إلى إلقاء نظرة على الأنماط الجينية على كروموسوم X واستبعاد المنطقتين PAR1 و PAR2 الموجودة في بدايتها ونهايتها. هذه هي الفواصل الزمنية للمواضع 60001-2699520 و 154931044-155260560 في نسخة GRCh37 من الجينوم. إذا كان التركيب الوراثي يحتوي على تسمية عددية واحدة ، فهذا هو الجنس البيولوجي للذكور ، وإلا فإن الأنثى. يجب أن يؤخذ في الاعتبار أن تعيين النوع الاجتماعي في ملف VCF يعتمد على توفر المعلومات حول المجال البيولوجي أثناء إنشاء VCF ، وبالتالي لا يمكن استخدام هذا النهج دائمًا.
استخدم الأمر التالي لكل من العينات في مجموعة البيانات. استبدال معرف العينة بعد الوسيطة
-s :
(/Data/Tutotrial/CEI.1kg.2019.demo.vcf.gz):
عند تنفيذ الأوامر ، سترى بعض محتويات ملف VCF لمعرف العينة المحدد.
-r chrX:2699521-154931043 في BCFtools عرض محتويات الملف إلى منطقة كروموسوم X من الموضع 2699521 إلى الموضع 154931043 (المنطقة غير PAR في الشكل 3). تستبعد هذه الحدود المناطق شبه الجسمية المزيفة غير الضرورية في هذه الحالة (PAR1 و PAR2). باستخدام القيم العددية في حقل GT ، حدد جنس كل عينة.
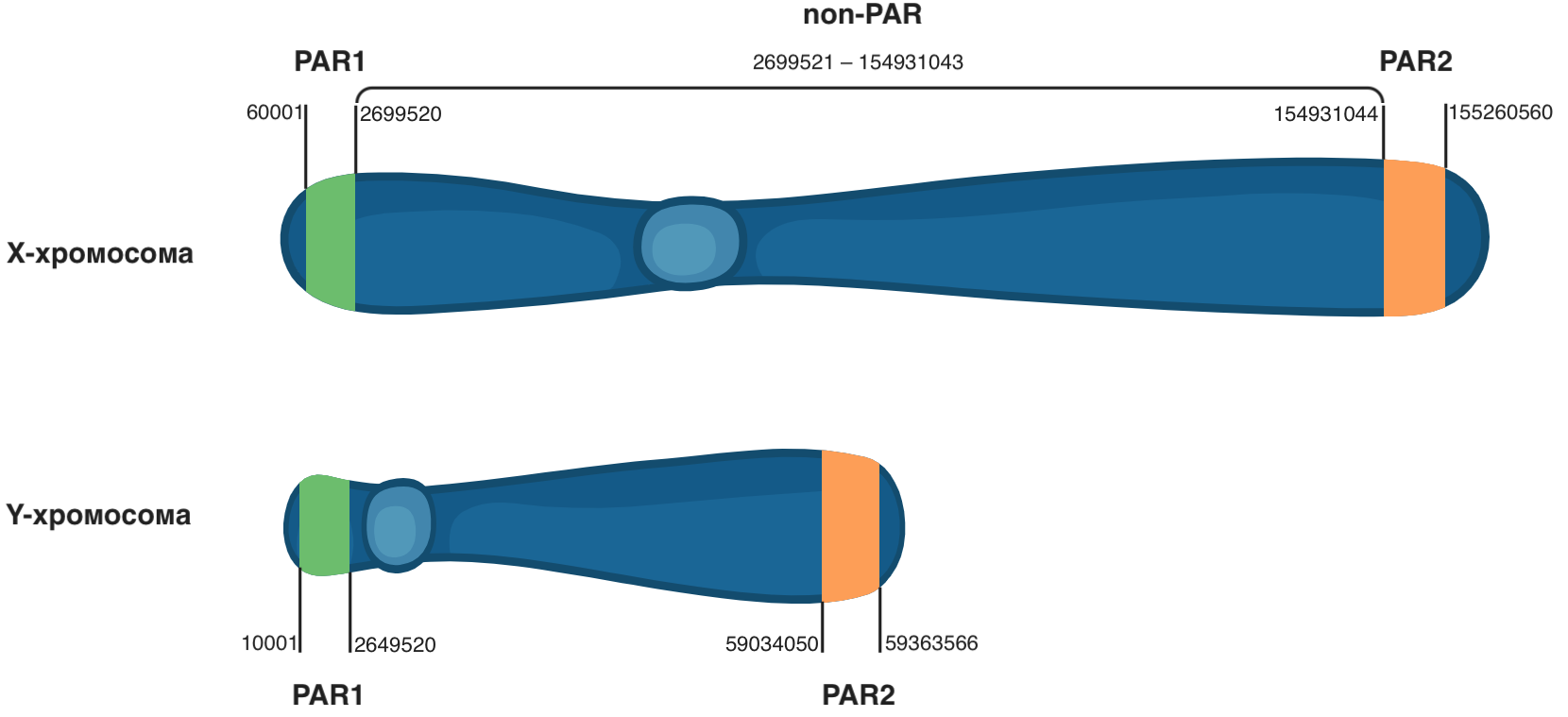 الشكل 3
الشكل 3 موقع مناطق شبه جسمية مزيفة من PAR1 و PAR2 على الكروموسومات الجنسية.
يمكنك الاطلاع على قائمة بجميع معرفات العينات في ملف VCF في الشكل 1 أو في السطر الأخير من رأس ملف VCF. سيتم سردها بعد اسم عمود FORMAT:
يظهر الجنس الحقيقي لهذه العينات أيضا في الشكل 1.
نحدد العلاقة
لتحديد العلاقة ، نحتاج إلى مقارنة البيانات الجينية لجميع العينات بشكل زوجي. من الصعب القيام بذلك وفقًا للجينوم الكامل: في هذه الحالة ، يأخذ ملف VCF عشرات الجيجابايت. يستغرق VCF الذي نستخدمه حوالي 2 غيغا بايت فقط ، لكننا ما زلنا نقوم بتصفية ذلك وفقًا لقائمة معرفات التباين الوراثي (rsIDs) الموروثة على الرقاقات من Illumina: GSA v1 ، GSA v2 ، HumanOmniExpress v1.0 ، HumanOmniExpress v1.3 ، InfiniumExome v1. 1 و Infinium OmniExpressExome v1.4. هذه هي رقائق الأكثر شعبية في التنميط الجيني التجاري.
قمنا بتجميع قائمة بجميع معرفات الاختلافات الجينية من هذه الرقائق في ملف منفصل مع قائمة rsIDs. أنه يحتوي على 1.4 مليون معرف. لتصفية ملف VCF ، قم بتشغيل الأمر التالي:
bcftools view -O z -i 'ID=@rsids_for_subsetting.txt' CEI.1kg.2019.demo.vcf.gz > CEI.1kg.2019.demo.subset.vcf.gz
في كل مرة تستخدم فيها BCFtools وحزم أخرى للعمل مع ملفات VCF ، تتم إضافة محفوظات الأوامر السابقة إلى رأس الملف. بغض النظر عن طريقة تصفية ملف VCF والأوامر المنفذة مسبقًا ، يمكنك التحقق من تكامل وهوية المحتويات الرئيسية لـ VCF عن طريق حساب مجموع التجزئة:
يقوم الأمر
gunzip -c ضغط الملف
gunzip -c محتوياته في stdout ، حيث يتم حذف خطوط رأس ملف VCF التي تبدأ
# (وبالتالي ، يتم استخدام الأمر
grep -v "^#" ). تتم إزالة الرأس من أجل مقارنة سلامة البيانات الوراثية نفسها فقط ، وليس البيانات الوصفية حول الأدوات ومتى تم استخدامها للعمل مع ملف VCF هذا.
إذا كانت قيمة التجزئة متطابقة ، فيمكنك الانتقال وتحويل VCF إلى تنسيق Plink الداخلي (بشكل افتراضي ، يكون تنسيق Plink هو ثلاثة ملفات تحتوي على ملحقات السرير و bim و fam). في هذه الملفات ، يبقى فقط التركيب الوراثي والكروموسوم والموضع وبعض البيانات الأخرى ، ويتم التخلص من الباقي. باستخدام هذا التنسيق ، أصبح العمل وحل المشكلات المختلفة التي لا تتطلب معلومات إضافية من VCF أسهل بكثير. على سبيل المثال ، قم بإجراء GWAS.
سيقوم هذا الأمر بإنشاء ثلاثة ملفات في المجلد:
CEI.1kg.2019.demo.subset.bed
CEI.1kg.2019.demo.subset.bim
CEI.1kg.2019.demo.subset.famيمكنك تحديد القرابة الزوجية لجميع العينات 10. نستخدم الأمر التالي لتحليل ملفات Plink:
king -b CEI.1kg.2019.demo.subset.bed --kinship --prefix CEI.1kg.2019.demo.subset.kinship_analysis
انظر إلى ملف
CEI.1kg.2019.demo.subset.kinship_analysis.kin0 إلى عمود القرابة ، الذي يحتوي على معاملات القرابة لأزواج العينات المشار إليها في ID1 و ID2 ، على التوالي.
قارن المعاملات التي حصلت عليها في الملف
CEI.1kg.2019.demo.subset.kinship_analysis.kin0 لجميع أزواج العينات مع النسب الموضح في الشكل 1 (الخط المتقطع يتوافق مع القرابة من الدرجة الثانية ، ومع ذلك ، لا توجد بيانات القرابة الدقيقة ، أي هذا قد يكون هناك أبناء عم أو عمة / ابن أخي أو عم / أخت). حاول أن تجعل استنتاجك الخاص حول قيم معاملات القرابة التي يمكن أن تتوافق مع ارتباط الدرجة الأولى والثانية.
مساعدةمقتطف من وثائق KING: معاملات القرابة> 0.354 تتوافق مع عينات مكررة أو توائم متطابقة ، من 0.177 إلى 0.354 إلى القرابة من الدرجة الأولى (أولياء الأمور - الأبناء ، الأشقاء) ، من 0.0884 إلى 0.177 إلى القرابة من الدرجة الثانية (أبناء العم ، العمات / الأعمام والأبناء) ، ومن 0.0442 إلى 0.0884 ، إلى القرابة من الدرجة الثالثة (الأجداد والأحفاد وأبناء العم الثاني). أي شيء أقل من 0.0442 يصعب تفسيره بشكل لا لبس فيه.
المهمة الأولى للمسابقة
باستخدام مجموعة بيانات اختبار من 12 عينة
Data/Test/CEI.1kg.2019.test.vcf.gz ،
Data/Test/CEI.1kg.2019.test.vcf.gz نسبهم ، مسترشدين بنتائج تحديد الجنس وتحليل القرابة. العينات التي ، وفقا لنتائج التحليل ، ليست في صلة القرابة مع شخص ما ، والكتابة في مكان قريب ، دون توصيلهم بخط مع عينات أخرى. يمكن تكوين النسب بنمط مشابه للشكل 1 ، ومع ذلك ، يبقى هذا وفقًا لتقديرك. يتم الإشارة إلى الرجال بواسطة مربع ، والمرأة على شكل دائرة ، والزواج بخط أفقي ، والطفل بخط عمودي ، والعديد من الأطفال بالتفرع الأفقي لخط عمودي (في شكل حرف P). اقرأ المزيد عن هذه التسميات
هنا .
كما ذكرنا أعلاه ، لا يمكن لمعاملات القرابة أن تميز بشكل واضح القرابة لأمر أو آخر: يتم الحصول على معاملات القرابة نفسها عند مقارنة الأزواج بين الوالدين والطفل والأخت الشقيقة (القرابة من الدرجة الأولى). إذا لم يكن من الممكن إثبات طبيعة العلاقة ، فقم بالإشارة إلى أي من الممكن. يرجى ملاحظة أن العينات الموجودة في مجموعة بيانات الاختبار تحتوي على معرفات مختلفة عن تلك المستخدمة في مجموعة بيانات التدريب.
يجب إرسال الردود على البريد
wgs@atlas.ru حتى 26 ديسمبر إلى 23:59. سيتم نشر مهمتين أخريين قريبًا ، وستظهر النتائج النهائية للمهام في 28 ديسمبر. سيحصل الفائز على اختبار الجينوم الكامل ، وسيحصل المركزان الثاني والثالث على اختبار الجين أطلس. سيكون هناك أيضًا جوائز خاصة من
Yandex.Cloud . لا يشارك موظفو Atlas السابقون والحاليون في المسابقة ؛)