نواصل دورة المهمة ، حيث نتحدث عن كيفية التعامل مع البيانات الجينية. يمكن بالفعل حل
المهمة الأولى "معرفة الجنس ودرجة العلاقة" وإرسال الإجابات إلينا. اليوم ننشر الثانية.
الجائزة الرئيسية هي
الجينوم الكامل .

لقد سبق أن شاركنا معلومات وروابط مفيدة قد تكون مفيدة للعمل مع بيانات المعلوماتية الحيوية. نوصيك بقراءة المقالات السابقة أولاً إذا فاتتك:
ما هو الجينوم الكامل ولماذا هو مطلوبالمهمة رقم 1. معرفة الجنس ودرجة العلاقة.تنصل
يتم العمل مع البيانات الوراثية على أنظمة Unix (Linux ، macOS) ، حيث أن بعض الأوامر والبرامج غير متوفرة على Windows. لذلك ، لمستخدمي Windows ، أحد أبسط الحلول هو استئجار جهاز Linux افتراضي.
يتم تنفيذ جميع العمليات الموضحة أدناه في سطر الأوامر - المحطة الطرفية. قبل البدء ، تعرف على كيفية العمل في محطة تشغيل نظام التشغيل الخاص بك واستخدام الأوامر ، لأن بعضها يمكن أن يضر نظام التشغيل والبيانات الخاصة بك.
البرامج المطلوبة
لقد جمعنا
صورة جهاز افتراضي (VM) مع جميع البرامج اللازمة على Yandex.Cloud. يمكن العثور على تعليمات إعداد VM وتثبيت البرنامج في
المقالة السابقة مع Task No. 1.
هذه المرة ، سوف تحتاج إلى بناء مخطط مبعثر ثنائي الأبعاد باستخدام البيانات التي تم الحصول عليها من خلال طريقة تحليل المكونات الرئيسية. نقترح إنشاء هذا المخطط باستخدام أي برنامج مناسب لك: Excel و Google Sheets و Python و R وغيرها.
لإكمال المهمة ، تحتاج إلى حزمة البرامج Plink 1.9. إذا لم تقم بتثبيته بعد (ولم تكمل المهمة رقم 1) ، فاقرأ المقالة السابقة. أنه يحتوي على تعليمات التثبيت. للمشاركة في مسابقة العام الجديد 2019 ، يجب إكمال جميع المهام!
يحيط علما
يعد تحليل المكون الرئيسي (PCA) أحد خوارزميات التعلم الآلي بدون المعلم عندما يبحث الجهاز بشكل مستقل عن أنماط البيانات. في علم الوراثة ، يسمح PCA بتجميع العينات وفقًا لبيانات التنميط الجيني في الفضاء ذي البعد N (عادة ثنائي الأبعاد) ، حيث تشرح المكونات الرئيسية التي تم الحصول عليها بدقة تباين البيانات الوراثية من عينة إلى عينة.
عند إجراء مثل هذا التحليل ، عادة ما تشكل عينات من مجموعة واحدة كتلة ، ويعتمد حجم وسلاسة الحدود التي تعتمد على تشابه العينات داخل مجتمع معين. من المحتمل أن تحدد الخوارزمية عينات من مجموعات مختلفة في مجموعات مختلفة. وسيتم تحديد العينات من المجموعات السكانية القريبة التي تنتمي إلى نفس السكان الفائقين ، على سبيل المثال ، EAS - شرق آسيا ، كما هو موضح في الشكل 1 ، بالقرب من بعضها البعض أو حتى في المجموعات المتقاطعة.
 الشكل 1
الشكل 1 نسب العينات المستخدمة في VCF (المربع يتوافق مع جنس الذكور ، الدائرة إلى الأنثى). الخط المتقطع يتوافق مع علاقة من الدرجة الثانية غير محددة.
يستخدم تحليل مماثل لتحديد السكان عن طريق التنميط الجيني. لهذا ، هناك حاجة إلى مجموعة بيانات مرجعية ، والتي تتكون من عينات ذات أصل معروف بالفعل. يمكن التوصل إلى استنتاج حول المجموعة التي تكون بها مجموعة من العينات المعروفة هي الأقرب إلى البيانات التي تمت دراستها.
ولتبسيط الأمر ، فإن جوهر تحليل PCA هو أن المسافات الزوجية بين النقاط في الفضاء متعدد الأبعاد معروفة ، ويجب أن تكون هذه النقاط موجودة في مساحة ذات بُعد أصغر بحيث تكون المسافات الزوجية الجديدة مختلفة إلى الحد الأدنى عن المسافات الأصلية. يعمل تقليل الأبعاد على تبسيط تحليل البيانات ، ولكن كلما قللنا ذلك ، كلما زادت المسافات الجديدة بين النقاط عن الأصل. لذلك ، تتضمن مهمة تحليل PCA أيضًا إيجاد حل وسط بين الدقة وسهولة التحليل. كل شيء كما في الحياة.
يعتمد أبسط تجسيد لتطبيق PCA على البيانات الوراثية على هوية بعض الأليلات ، والتي يمكن تقسيمها إلى نوعين فرعيين: IBS (الهوية حسب الولاية) و IBD (الهوية حسب النسب). يعني IBS هوية بعض الأليلات في شخصين ، ولكن لا يعني بالضرورة وجود أي علاقة بينهما. على العكس من ذلك ، يتحدث IBD عن هوية الأليلات بسبب وجود سلف مشترك ، وبالتالي صلة قرابة.
أليلات IBD هي أليلات IBS بشكل لا لبس فيه ، في حين أن العكس ليس صحيحًا. ومع ذلك ، يجب أن يؤخذ في الاعتبار أننا في وقت ما قد أتينا من سلف مشترك ، لذلك قد تكون بعض الأليلات IBD. في تحليل PCA أدناه ، يتم استخدام مفهوم IBS فقط ، على الرغم من أنه في التحليلات الأكثر تعقيدًا ، يأخذ في الاعتبار اختبارات الأهمية الإحصائية والقيود المظهرية وأحجام المجموعات والعمر والجنس للشخص بالإضافة إلى معلومات إضافية حول بنية السكان.
وكلما زاد عدد الأليلات المختلفة في عينتين ، قل عدد التشابه والأبعد عن بعضها البعض. ستكون قيمة IBS لهذه العينات منخفضة. ولكن بالنسبة للوالدين وأطفالهم ، فإن IBS سيكون مرتفعًا جدًا.
معرفة قيم IBS لكل زوج من الصور في مجموعة البيانات ، يمكنك إجراء تحليل PCA لمعرفة كيف يتم تجميعها.
يستخدم الاختبار الوراثي أطلس خوارزمية أكثر تطوراً لتحديد تمثيل السكان في بيانات التنميط الجيني.
البيانات المستخدمة
نذكرك أن الدليل يستخدم البيانات المفتوحة المحددة بشكل خاص من مشروع
1000 Genomes . للتحليل ، تم اختيار 10 عينات تحتوي على معلومات عن التركيب الوراثي من حوالي 85 مليون تنوع ، والتي تم الحصول عليها من خلال تحليل بيانات NGS المتوافقة مع نسخة الجينوم GRCh37. وتظهر العلاقات الأسرية والسكان من هذه العينات في الشكل 1.
بناء التجمعات السكانية
استخدم الملفات الثلاثة بتنسيق Plink الذي تم الحصول عليه مسبقًا في المهمة رقم 1:
CEI.1kg.2019.demo.subset.bed CEI.1kg.2019.demo.subset.bim CEI.1kg.2019.demo.subset.fam
تحديد المسافة الزوجية بين جميع العينات 10 في مجموعة بيانات التدريب ورسم PCA على أساس IBS (الهوية من قبل الدولة). يمكن القيام بذلك على النحو التالي:
المعلمة
—genome هي المسؤولة فقط عن حساب
—genome IBS / IBD بين جميع العينات في مجموعة البيانات. المعلمة "
—read-genome " هي مصفوفة المسافة الزوجية التي تم الحصول عليها في وقت سابق ، والمعلمات "
—cluster —mds-plot 10 هي المسؤولة عن تحليل PCA وإخراج نتائجها إلى جدول المكونات الرئيسية العشرة الأولى. في الواقع ، هذه هي إحداثيات كل عينة في الفضاء 10 الأبعاد.
سيُنشئ الأمر الأخير 4 ملفات في المجلد:
CEI.1kg.2019.demo.subset.clustering.cluster1 CEI.1kg.2019.demo.subset.clustering.cluster2 CEI.1kg.2019.demo.subset.clustering.cluster3 CEI.1kg.2019.demo.subset.clustering.mds
سنحتاج إلى آخر ملفين من القائمة.
يوضح الشكل 2 كيف يبدو الملف الذي تم استلامه في مجموعة بيانات تدريب MDS. تتوافق حقول FID (معرّف العائلة) و IID (معرّف فردي) مع معرّفات العينة الفردية والعائلة. يحتوي الحقلان C1 - C10 على قيم كل مكون من المكونات الرئيسية العشرة لكل عينة ، حيث يشرح المكون C1 إلى الحد الأقصى تباين البيانات الوراثية للعينات التي تم تحليلها ، و C10 على أقل تقدير.
 الشكل 2
الشكل 2 ملف MDS بقيم 10 مكونات رئيسية لكل عينة.
عند إنشاء مخطط مبعثر باستخدام مكونين (في مساحة ثنائية الأبعاد) ، يمكن للمرء اكتشاف مجموعات تتوافق مع مجتمع العينة. يوضح الشكل 3 مخططات التشتت لأزواج المكونات الرئيسية C1xC2 و C2xC3 و C1xC3. عند مقارنة المجموعات التي تم الحصول عليها مع الانتماء المرجعي للسكان (الشكل 1) ، يُظهر زوج المكونين الأولين C1 - C2 أعلى دقة (100٪) ، ويفصل بشكل صحيح جميع العينات وفقًا لانتماء سكانها المعلن في مشروع 1000 Genomes. ومع ذلك ، فمن المنطقي دائمًا مقارنة النتائج التي تم الحصول عليها لعدة أزواج من المكونات بسبب التداخل أو الفصل المحتمل للمجموعات الحقيقية.
 الشكل 3
الشكل 3 قطع مبعثر لمواقع العينات لأزواج من المكونات الرئيسية ؛ تم تغيير موقع العلامات قليلاً لمنع تداخلها.
يمكن أن يساعد أيضًا بناء المخططات ثلاثية الأبعاد باستخدام المكونات الثلاثة الرئيسية الأولى في تحديد المجموعات ، ولكن ليس دائمًا. على سبيل المثال ، يتيح لنا إنشاء مثل هذا الرسم البياني للبيانات في الشكل 3 تحديد 4 مجموعات يتم فيها تجميع عينات من مجموعات PUR و KHV وفقًا للمجموعات السكانية ، وتنقسم العينات من سكان CDX إلى مجموعتين (الشكل 4). هذا هو أيضا ملحوظ في الشكل 3 في الإحداثيات C2xC3 و C1xC3.
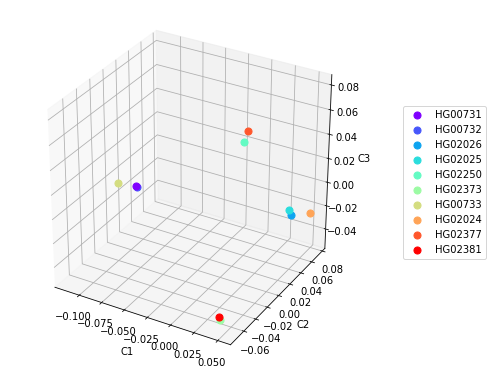 الشكل 4
الشكل 4 مؤامرات مبعثر للمكونات الرئيسية الثلاثة.
يمكن تفسير هذه النتائج المتضاربة للتحليل بعدد صغير من العينات ، نظرًا لأن قيم المكونات الرئيسية لكل عينة مختلفة عن مجموعات البيانات المختلفة في الحجم والتكوين ، وعندما يتم تضمين عينات إضافية من مجموعات مختلفة ، قد تتغير نتيجة التجميع. من المحتمل أيضًا حدوث أخطاء عند إنشاء مجموعة بيانات وتوفير بيانات مرجعية عن مجموعة العينات ، ومع ذلك ، في مشروع 1000 Genomes ، يكون احتمال حدوث مثل هذا الموقف منخفضًا للغاية.
لا يستخدم ملف MDS علامات تبويب أو فواصل كمحددات ، لذلك اضبط تنسيقه للراحة. استخدم
tab أو ملف
csv كوسيطة ثانية:
سيقوم الفريق بإنشاء ملف
CEI.1kg.2019.demo.subset.clustering.mds.tab ، والذي يمكنك تنزيله وإنشاء مخططات مبعثرة مماثلة لتلك الموضحة في الشكل 3. مقارنة النتائج ، يجب أن تكون متطابقة مع تلك المشار إليها أعلاه.
بناء شجرة التجميع
يمكنك أيضًا تقييم تجميع العينات باستخدام شجرة ثنائية ، والتي تمثل معلومات المجموعات حول العينات في شكل منفصل. توجد معلومات حول هذه الشجرة في ملف
CEI.1kg.2019.demo.subset.clustering.cluster3 (الشكل 5).
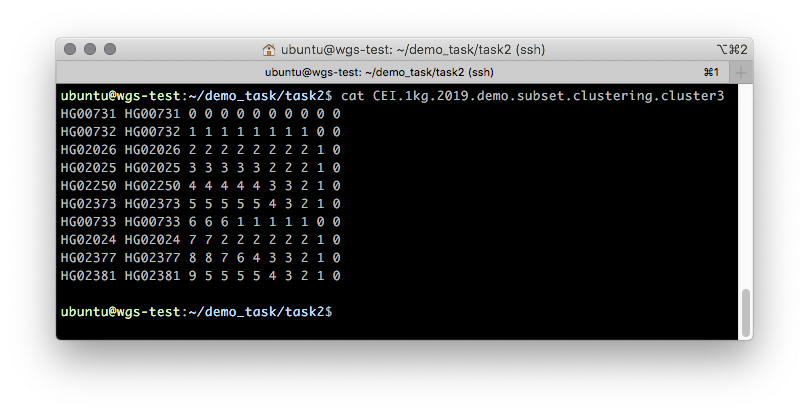 الشكل 5
الشكل 5 المحتويات
.cluster3 لملف
.cluster3 الذي يصف عملية التجميع التدريجي للعينات من مجموعة واحدة إلى N ، حيث N هو عدد العينات.
يحتوي أول عمودين من هذا الملف على FID و IID. يوصف الانتماء العنقودي من قبل الجميع. يجب قراءة هذا الملف من اليمين إلى اليسار في أعمدة بزيادات عمود واحد: في البداية ، تنتمي جميع العينات إلى مجموعة واحدة "0" - العمود أقصى اليمين. عند تقسيمها إلى مجموعتين (في الخطوة الثانية ، في العمود الثاني) ، تظهر مجموعتان: "0" و "1" ، حيث تحتوي المجموعة "0" على عينات HG00731 و HG00732 و HG00733 ، بينما تحتوي المجموعة "1" على الباقي. يظهر الشكل التوضيحي لهذا القسم في الشكل 6.
من الشجرة ، يمكن أن نستنتج أن العينات تنتمي إلى السكان (الشكل 1). بالإضافة إلى ذلك ، يتيح لنا بناء هذه الشجرة تحديد القرب من السكان الأفراد ، أي حدوث تجمعات CDX و KHV في تجمع سكاني واحد EAS (بالفعل في الخطوة الأولى لتقسيم المجموعات السكانية الفائقة يتم فصل EAS و AMR في فرعين حاليين). أيضا ، يمكن أن يساعد بناء شجرة التجميع في تصحيح النتائج الغامضة لتصور العينات للمكونات الرئيسية.
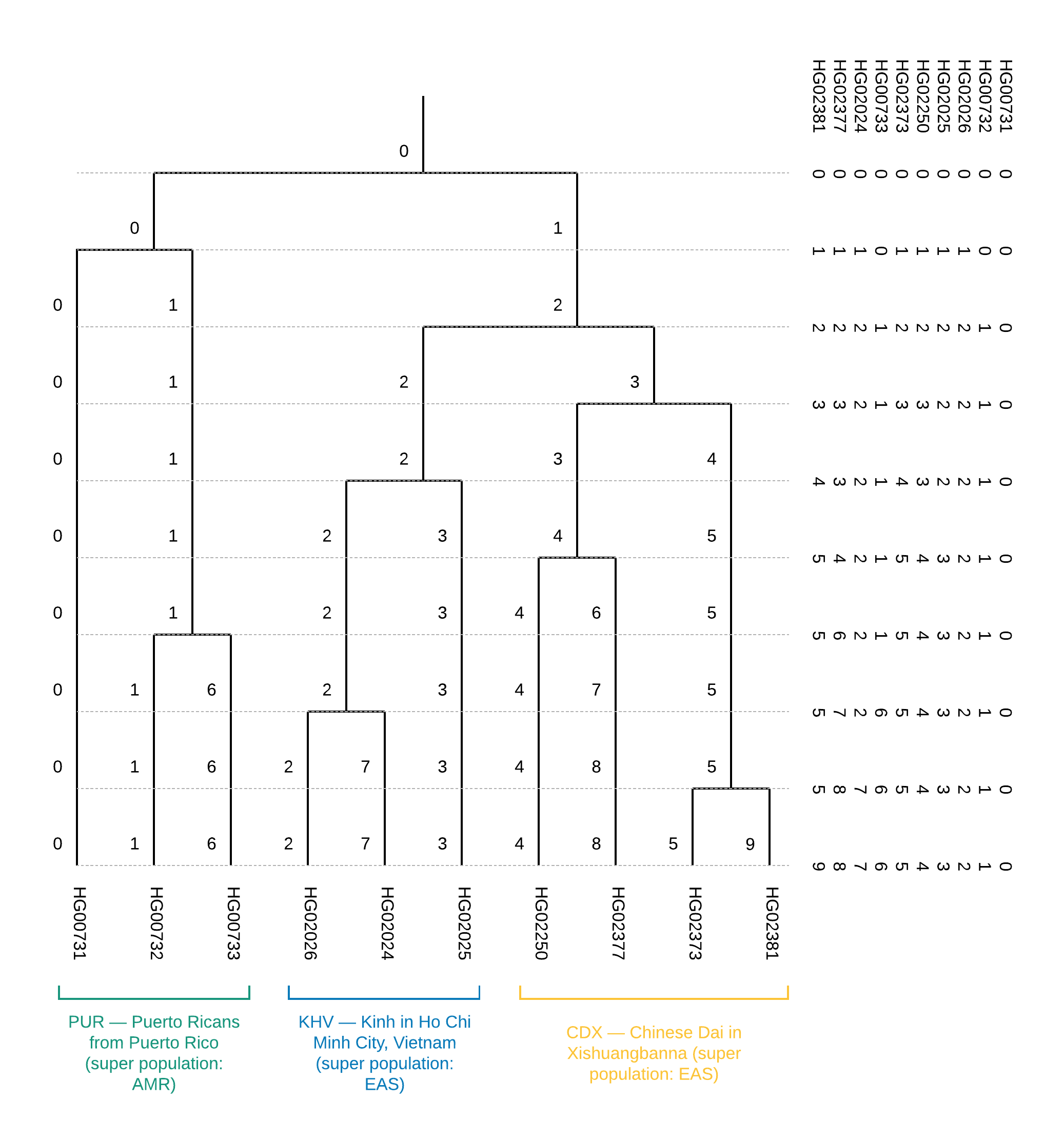 الشكل 6
الشكل 6 شجرة التجميع الثنائية لمجموعة بيانات التدريب من 10 عينات: على اليمين هو محتويات الملف
CEI.1kg.2019.demo.subset.clustering.cluster3 (من اليمين إلى اليسار في الملف ، بشكل مماثل من أعلى إلى أسفل في الشكل).
المهمة الثانية للمسابقة
استخدم مجموعة بيانات الاختبار من 12 عينة من
Data/Test/CEI.1kg.2019.test.vcf.gz والمثال أعلاه (الشكل 5) لإنشاء شجرة تجميع ثنائية من ملف
.cluster3 الذي
.cluster3 وإرفاقه بالمحلول. قم بتحليل الشجرة الناتجة واستخلص استنتاجات حول عدد المجموعات السكانية الكبيرة المقدمة في مجموعة بيانات الاختبار.
حدد المجموعات السكانية المكونة من 12 عينة من مجموعة بيانات الاختبار من خلال تحليل المكونات الرئيسية C1 و C2 و C3 مع مراعاة الشجرة المبنية وتشير إلى ذلك على النسب المبنية في المشكلة رقم 1 ، وتقييد كتل السكان الفردية (على غرار الشكل 1). يجب أن توضع العينات التي لم تظهر وجود القرابة في المشكلة رقم 1 بالطريقة نفسها داخل الكتل التي تم الحصول عليها في الرسم البياني دون ربطها بخطوط مع عينات أخرى. لا تنسى إرفاق المؤامرات المبعثرة التي قمت بإنشائها.
يجب إرسال الردود على البريد
wgs@atlas.ru حتى 26 ديسمبر إلى 23:59. سيتم نشر مهمة أخرى قريبًا ، وستظهر النتائج النهائية للمهام في 28 ديسمبر. سيحصل الفائز على اختبار الجينوم الكامل ، وسيحصل المركزان الثاني والثالث على اختبار الجين أطلس. سيكون هناك أيضًا جوائز خاصة من
Yandex.Cloud . لا يشارك موظفو Atlas السابقون والحاليون في المسابقة ؛)