ننشر اليوم المهمة الأخيرة من الدورة التي نحكي فيها كيفية التعامل مع البيانات الجينية. تم نشر المهام
الأولى والثانية بالفعل: يمكن حلها وإرسال الإجابات إلينا. نحن نحذرك من أن هذه المهمة تستغرق وقتًا أطول من الباقي.
الجائزة الرئيسية هي
الجينوم الكامل .

لقد سبق أن شاركنا معلومات وروابط مفيدة قد تكون مفيدة للعمل مع بيانات المعلوماتية الحيوية. نوصيك بقراءة المقالات السابقة أولاً إذا فاتتك:
ما هو الجينوم الكامل ولماذا هو مطلوبالمهمة رقم 1. معرفة الجنس ودرجة العلاقة.المهمة رقم 2. تحديد التركيبة السكانيةتنصليتم العمل مع البيانات الوراثية على أنظمة Unix (Linux ، macOS) ، حيث أن بعض الأوامر والبرامج غير متوفرة على Windows. لذلك ، لمستخدمي Windows ، أحد أبسط الحلول هو استئجار جهاز Linux افتراضي.
يتم تنفيذ جميع العمليات الموضحة أدناه في سطر الأوامر - المحطة الطرفية. قبل البدء ، تعرف على كيفية العمل في محطة تشغيل نظام التشغيل الخاص بك واستخدام الأوامر ، لأن بعضها يمكن أن يضر نظام التشغيل والبيانات الخاصة بك.
البرامج المطلوبة
لقد جمعنا
صورة جهاز افتراضي (VM) مع جميع البرامج اللازمة على Yandex.Cloud. يمكن العثور على إرشادات إعداد VM وتثبيت البرنامج في
المقالة مع المهمة الأولى. هناك أيضًا إرشادات حول كيفية إعداد الجهاز لاستخدامه مجانًا حتى 31 كانون الأول (ديسمبر) 2019.
في هذه المهمة ، تحتاج إلى تحويل بيانات التنميط الجيني من تنسيق VCF إلى تنسيق 23andMe ، وتحميل الملفات المستلمة إلى خدمة Promethease والتعرف على محتويات التقرير لكل عينة.
تنسيق 23andMe هو تنسيق نصي لتخزين بيانات التنميط الجيني ويحتوي على 4 حقول مفصولة بعلامات جدولة. يحتوي الحقل الأول على معرف التباين (على سبيل المثال ، rsID) ، والثاني يحتوي على كروموسوم (القيم الصالحة لهذا الحقل هي 1-22 و X و Y و MT) ، بينما يحتوي الثالث على الموضع على كروموسوم ، بينما يحتوي الرابع على النمط الوراثي (ثنائي الصبغيات في وجود اثنين من الكروموسومات المتجانسة ، والمفرد في الآخرين الحالات). هذا التنسيق مدعوم من قبل العديد من خدمات الترجمة الفورية ، لذلك في المهمة التي سنعمل بها.
لإكمال المهمة ، تحتاج إلى حزمة البرامج BCFtools. إذا لم تقم بتثبيته بعد ، فاقرأ
المقالة مع المهمة الأولى. أنه يحتوي على تعليمات التثبيت. نذكرك بأنه للمشاركة في مسابقة السنة الجديدة 2019 ، يجب إكمال جميع المهام.
بالإضافة إلى BCFtools ، ستحتاج إلى
create_23andme.sh وهو برنامج نصي bash يُستخدم لإنشاء البيانات بتنسيق 23andMe. يوجد هذا الملف في الدليل
/Technical على Yandex.Cloud وكذلك في الأرشيف للتنزيل ، وهو متاح عبر الرابط في
المقالة .
يحيط علما
هناك العديد من الخدمات التي تقوم بتحليل بيانات التنميط الجيني: MyHeritage و Promethease و FamilyTreeDNA و DNA.LAND و GEDmatch. أنها توفر تنزيل بيانات التنميط الجيني في أشكال مختلفة ، وغالبا ما تكون محددة لموفر التنميط الجيني معين (Ancestry ، 23andMe ، MyHeritage ، FamilyTreeDNA ، GenesForGood وغيرها). الأكثر ولاءً لتنسيق البيانات هو Promethease: يمكنك تنزيل كل من ملفات VCF و 23andMe في هذه الخدمة.
هناك العديد من مشكلات التوافق بين التنسيقات والخدمات:
- تستخدم الشركات المختلفة إصدارات مختلفة من الجينوم لرسم خريطة للتغيرات الجينية ، ويتم حل هذه المشكلة من خلال ما يسمى بالرفع ، عندما يتم استبدال مواقع الاختلافات الجينية في البيانات المصدر بأخرى مقابلة في نسخة أخرى من الجينوم. على سبيل المثال ، يوفر Atlas بيانات التنميط الجيني لإصدار جينوم GRCh38 ، ويتلقى GEDmatch بيانات للإصدار السابق من جينوم GRCh37. يسمى تحويل إحداثيات التغيرات الجينية من GRCh38 إلى GRCh37 المصعد.
- استخدام معرفات فريدة للتغيرات الجينية بخلاف rsIDs. يتم حل عدم التوافق هذا عن طريق استبعاد هذه الإدخالات من الملف أو التعليق عليها بتخصيص rsID. والثاني ليس ممكنًا دائمًا.
- تستخدم الخدمات مجموعة ثابتة من الاختلافات الجينية. في بعض الأحيان يؤدي عدم تطابق جزء على الأقل من البيانات التي يتم تنزيلها إلى حدوث خطأ في التحميل. هذه المشكلة ذات صلة ، على سبيل المثال ، MyHeritage. يمكن حلها عن طريق تسليط الضوء على مجموعة من معرفات الاختلافات الجينية التي لا تسبب خطأ في التحميل.
البيانات المستخدمة
نذكرك أن هذا الدليل يستخدم البيانات المفتوحة المختارة خصيصًا من مشروع
1000 Genomes . للتحليل ، اخترنا 10 عينات مع معلومات التركيب الوراثي من ~ 85 مليون الاختلافات ، التي تم الحصول عليها من خلال تحليل بيانات NGS محاذاة مع نسخة من الجينوم GRCh37. وتظهر العلاقات الأسرية والسكان من هذه العينات في الشكل 1.
 الشكل 1
الشكل 1 نسب العينات المستخدمة في VCF (المربع يتوافق مع جنس الذكور ، الدائرة إلى الأنثى). الخط المتقطع يتوافق مع علاقة من الدرجة الثانية غير محددة.
تحويل VCF
فيما يلي إرشادات حول تحويل ملف VCF وتحميل البيانات المستلمة إلى خدمة Promethease ، والتي أصبحت مؤخرًا مجانية. نقترح عليك أن تتعرف على تقرير Promethease الذي تم استلامه في أي من العينات. استخدم ملف VCF الذي تمت تصفيته بواسطة قائمة الاختلافات التي تم الحصول عليها في
المهمة رقم 1 .
يسمح لك الأمر
bcftools query باستخراج أي معلومات متاحة بتنسيق محدد بواسطة المستخدم بعد علامة
-f من ملف VCF. تشير علامة
-s إلى معرف العينة (
HG00731 ) لاستخراج البيانات. يتم استخدام علامة -e للإشارة إلى معايير الاستبعاد ، في هذه الحالة
'%ID=="."' باستثناء الإدخالات التي لا تحتوي على rsID. يتم تمرير الإخراج من
bcftools query إلى البرنامج النصي
create_23andme.sh ، الذي يحول البيانات إلى تنسيق TSV بأربعة أعمدة (rsID ، كروموسوم ، موضع ، نمط جيني) ويكتبها في ملف. يمكنك تنزيل وحفظ البرنامج النصي
create_23andme.sh لنفسك للعمل مع بيانات التسلسل الكامل للجينوم الخاصة بك.
يستخدم البرنامج النصي
create_23andme.sh المستخرجة من ملف VCF لتحديد نوع التباين الوراثي (الاختلاف أحادي النوكليوتيدات في SNV أو إدخال INS أو حذف DEL) وكتابة المعرّف rsID والكروموسوم والموضع والأليلات في
stdout وفقًا لنوع التباين المحدد (A ، G ، T و C هما أليلتان صالحتان لنوع SNV و I و D هما تسميات أليل صالحة لأنواع INS و DEL).
ضع في اعتبارك أن عملية التحويل تستغرق الكثير من الوقت: حوالي 4 ساعات لكل ملف لعينة واحدة مع ما يقرب من مليون صيغة. التزامن BCFtools غير معتمد.
الذهاب إلى
promethease.com والتسجيل. انقر فوق الزر تحميل البيانات الأولية (الشكل 2) وقم بتحميل الملف
HG00731.subset.23andme.txt . بعد اكتمال التنزيل ، انقر فوق الزر "إنشاء تقرير مجاني" وأدخل الاسم المطلوب للتقرير الذي سيتم إنشاؤه وفقًا لبياناتك. بعد إعداد التقرير ، ستتلقى إشعارًا عبر البريد الإلكتروني ويمكنك التعرف على محتويات التقرير. في تقارير كل عينة ، ابحث عن فصيلة الدم المحددة بواسطة نظام تفسير Promethease في نظام AB0 / Rh (عامل Rh - Rh). تحقق نتائجك للامتثال للجدول 1.
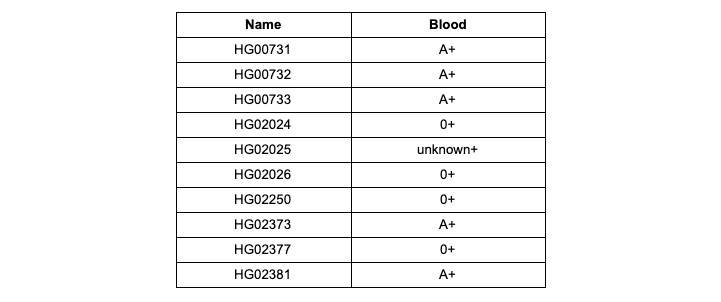 الجدول 1
الجدول 1 . فصائل الدم وعامل Rh تم الحصول عليها من تحليل Promethease للعينات من مجموعة البيانات التجريبية
يستخدم الأطلس عتبات تختلف عن Promethease لإدراج سمة معينة في التفسير حسب مستوى الأدلة. يشير مستوى الأدلة إلى مجمل نتائج الاختبارات والمعايير الإحصائية لأهمية كل علاقة تمت ملاحظتها بين التباين الوراثي وأي ميزة في جسم الإنسان. تحتوي العديد من السمات التي يمكن العثور عليها في تقرير Promethease على مستوى منخفض من الأدلة و / أو مستوى عالٍ فقط في مجموعة محدودة من السكان ، على سبيل المثال فقط لممثلي السكان الآسيويين.
مذكرةمن الناحية التجريبية ، قمنا بتثبيت قائمة من الاختلافات الوراثية استنادًا إلى شريحة
Infinium Global Screening Array v2.0 التي يمكن تحميلها على MyHeritage. يتم تخزين هذه القائمة (
external_interpretation_rsids.txt ) في ملف منفصل في الدليل
/Technical ، ويمكن استخدامها لتصفية VCF مع التحويل اللاحق عن طريق القياس مع الإرشادات المذكورة أعلاه. يمكنك أيضًا استخدام هذا الملف لتصفية بيانات التنميط الجيني من شريحة بحيث يمكنك تحميلها على MyHeritage. إذا كان لديك بالفعل اختبار الوراثة أطلس ، فيمكنك تحميل بيانات التنميط الجيني بتنسيق من حسابك الشخصي وتصفيته وفقًا لقائمة الاختلافات المقترحة - العمود الأول في البيانات التي تم تحميلها من حسابك الشخصي.
لاحظ أن الملفات المستخدمة في هذا الدليل تحتوي دائمًا على حقل ALT معبأ (أليل بديل) ، مما يجعل من الممكن فهم النوع الذي ينتمي إليه كل اختلاف (INS ، DEL ، SNV) وإنشاء إدخال بتنسيق 23andMe بشكل صحيح. تحتوي بيانات التسلسل على مستوى الجينوم في Atlas على أليل ALT المعبأ فقط في الأماكن التي تم اكتشاف هذا الأليل فيها ، وإلا فإن المعلومات الخاصة بملء حقل ALT عند إنشاء ملف VCF ببساطة غير موجودة. من الضروري إخراج البيانات في المواقع المرجعية المتجانسة (المواقف في الجينوم حيث لم يتم العثور على أليل مرجعي) ، لأن ليس فقط الاختلافات المكتشفة في تسلسل النيوكليوتيدات ، ولكن أيضًا لغيابها تأثير سريري.
عدم وجود أليل ALT في مثل هذه المواقف من الجينوم لا يسمح لنا بتحديد نوع التباين الوراثي الذي تم العثور على أليل الإشارة (REF) عليه فقط. تسجيل الأنماط الجينية لمثل هذه الحالات معقد بسبب الحاجة إلى استخدام مصدر للمعلومات حول الأليلات المحتملة لهذا الاختلاف ولا يشملها هذا الدليل. إذا كان من المحتمل أن تستخدم هذا الدليل
create_23andme.sh النصي
create_23andme.sh لتحويل ملف VCF الذي تم الحصول عليه بعد التسلسل على نطاق الجينوم إلى Atlas ، فلن يحتوي الملف المحول على أنماط وراثية متماثلة
create_23andme.sh ، لأن البرنامج النصي
create_23andme.sh بتصفية هذه السجلات بشكل صريح للقضاء على الأخطاء عند إنشاء سجلات للإدراج والحذف.
لكي لا يزال النص البرمجي
create_23andme.sh ينتج عن الأنماط الوراثية
create_23andme.sh المرجعية ، تحتاج إلى استبدال محتويات الأسطر 25-28 فيه
... if [ "$ALT" == "." ] || [[ "$ALT" == *"*"* ]] then continue fi ...
في
... if [[ "$ALT" == *"*"* ]] then continue fi if [ "$ALT" == "." ] then echo -e "$RSID\t$CHR\t$POS\t$REF$REF" fi ...
يسمح هذا الاستبدال بعرض إدخالات
stdout مع أنماط وراثية مرجعية متماثلة اللواقح. يجب أن يؤخذ في الاعتبار أن مثل هذه الإدخالات الخاصة بالإدخالات والحذف ستكون غير صحيحة ، لأن الأليلات الصحيحة في التنسيق المستخدم في عمليات الإدراج والحذف هي I و D ، وسيستخدم البرنامج النصي أليلات A أو G أو T أو C. الإدخالات والحذف ، من الضروري أن نتعرف مسبقًا على نوع الاختلاف الذي يميز موضع معين للجينوم الذي لم يتم فيه اكتشاف أليل ALT. يمكن الحصول على هذه المعلومات من خلال تحليل أليل ALT إذا كان متاحًا (مطبق بالفعل في
create_23andme.sh ) أو باستخدام قاعدة بيانات خارجية ، على سبيل المثال ، dbSNP (وليس في
create_23andme.sh ).
من أجل الحصول على تقرير Promethease عن ملف VCF كامل من تسلسل الجينوم الكامل في Atlas ، يمكنك تحميل ملف VCF نفسه في Promethease ، ومع ذلك ، عليك أن تضع في اعتبارك أن حجم ملف Atlas VCF المضغوط يبلغ حوالي 8 غيغابايت ، بينما يسمح لك Promethease بتحميل الملفات أكثر من 4 غيغا بايت. وصف الحلول لهذه المشكلة متوفر
هنا . الحل الآخر هو تقسيم ملف VCF إلى عدة أجزاء (أقل من 4 غيغابايت لكل منهما) وتحميل كل منها كملف إضافي في قائمة تنزيل بيانات Promethease.
المهمة الثالثة للمنافسة
قم بتنزيل البيانات المحولة لكل من العينات الـ 12 من مجموعة بيانات الاختبار ، والتي قمت بتصفيةها وفقًا لقائمة الاختلافات في المهمة الأولى ، في Promethease وقم بتجميع جدول مراسلات لمعرّف العينة - مجموعة الدم AB0 / Rh المعرّفة من قِبل نظام تفسير Promethease (عامل Rh - Rh). حددت فصائل الدم بشكل احتمالي وسجلت مع البادئة "prob" في تقرير Promethease ، اكتب بدون البادئة. سجل قيمًا غير محددة على أنها غير معروفة (لا يزال هناك حاجة إلى كتابة Rhesus factor لمجموعات الدم غير المعروفة ، إذا تم تعريفها). ويرد مثال في الجدول 1.
إن تحويل VCF إلى التنسيق المستخدم أعلاه في التطبيق المقترح مبسط إلى حد كبير ، ولكنه يتطلب قدراً كبيراً من الوقت. للتحسين ، يمكنك كتابة نص برمجي مع حلقة من شأنها أن تولد هذه البيانات تلقائيًا ، متكررة على مجموعة من المعرفات. من الممكن إجراء العديد من هذه البرامج النصية ، ولكل نقل مجموعات مختلفة من معرفات العينات للتنفيذ المتوازي ، ومع ذلك ، يجب ألا يتجاوز عدد البرامج النصية قيد التشغيل المتوازي عدد وحدة المعالجة المركزية لجهاز الكمبيوتر / الجهاز الظاهري. يتوفر وصف جيد لإنشاء مثل هذه الحلقات
هنا . عند العمل على Yandex.Cloud ، يمكنك ، إذا لزم الأمر ، إنشاء جهاز ظاهري آخر مع عدد كبير من وحدات المعالجة المركزية الافتراضية ، مما يقلل بشكل متناسب الوقت الذي يستغرقه إكمال المهمة.
هذه هي المهمة الأخيرة لدورتنا.
يجب إرسال الردود على البريد
wgs@atlas.ru حتى 26 ديسمبر إلى 23:59. سننشر الإجابات الصحيحة وأسماء الفائزين في 28 ديسمبر. سيحصل الفائز على اختبار الجينوم الكامل ، وسيحصل المركزان الثاني والثالث على اختبار الجين أطلس. سيكون هناك أيضًا جوائز خاصة من
Yandex.Cloud . لا يشارك موظفو Atlas السابقون والحاليون في المسابقة ؛)