Als der britische Morphologe George Jackson Mywart [St. George Jackson Mivart] veröffentlichte 1865 einen der ersten Evolutionsbäume, ihm fehlte unterstützendes Material. Er baute einen Baum - eine
verzweigte Karte verschiedener Primatenarten - unter Verwendung einer detaillierten Analyse der Tierstacheln. Der zweite Baum, der auf einem Vergleich der tierischen Gliedmaßen basiert,
zeigte andere familiäre Bindungen zwischen Primaten und hob das bis heute bestehende Problem der Evolutionsbiologie hervor.
Fast 150 Jahre später erhielten Wissenschaftler Datenberge, um die sogenannten
phylogenetischen Bäume zu bauen, eine moderne Version der von Mivart geschaffenen Struktur. Dank der Fortschritte in der DNA-Dekodierungstechnologie und der
Bioinformatik können Sie die Sequenzen von Hunderten von Genen und manchmal ganzen Genomen verschiedener Arten vergleichen und den Lebensbaum detaillierter als je zuvor erstellen.
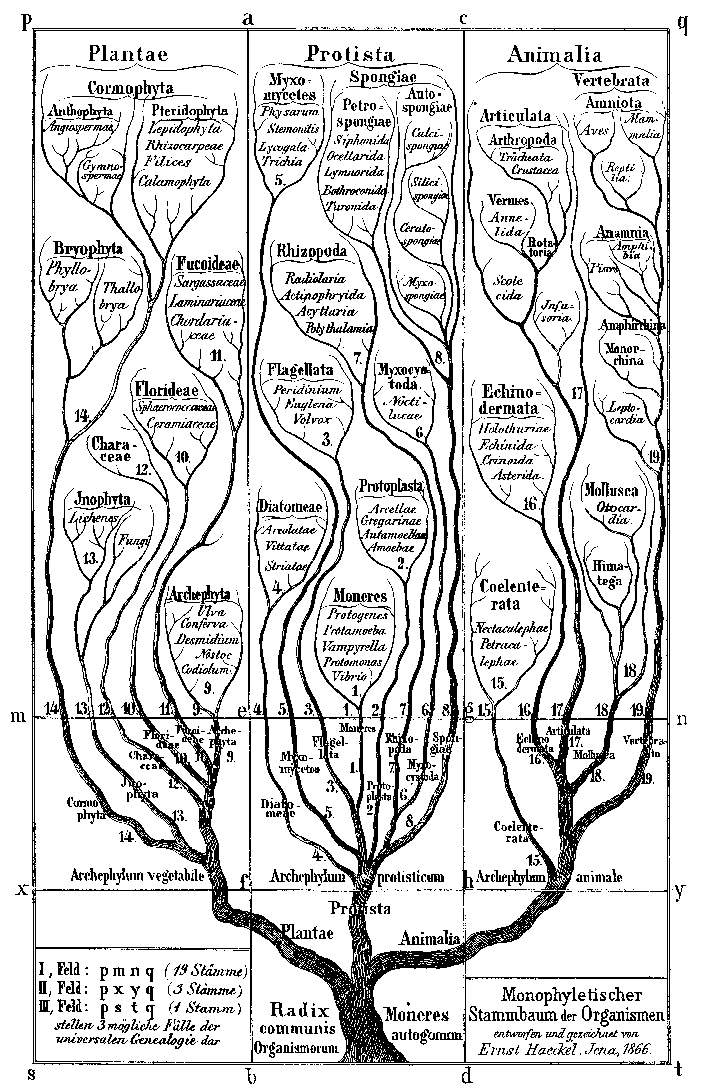 Der historische Lebensbaum von 1866 beschreibt die Königreiche der Pflanzen, Tiere und einzelligen
Der historische Lebensbaum von 1866 beschreibt die Königreiche der Pflanzen, Tiere und einzelligenObwohl die Fülle an Daten dazu beitrug, einige der Konflikte zu lösen, die über verschiedene Teile des Evolutionsbaums entstanden waren, brachte dies auch neue Schwierigkeiten mit sich. Die heutige Version des Baumes des Lebens ähnelt eher einer kontroversen Wikipedia-Seite als einem veröffentlichten Buch - es gibt laufende Debatten über einige Zweige. So wie Wirbelsäule und Gliedmaßen zu widersprüchlichen Karten der Evolution von Primaten führten, wissen Wissenschaftler jetzt, dass verschiedene Gene im selben Organismus unterschiedliche Geschichten erzählen können.
Laut einer neuen Studie, die teilweise auf der Untersuchung von Hefen basiert, ist das kontroverse Bild einzelner Gene noch widersprüchlicher als erwartet. "Es wird behauptet, dass jedes der 1070 Gene in einen Konflikt verwickelt ist", sagt Michael
Donoghue , ein Evolutionsbiologe in Yale, der nicht mit der Studie verbunden ist. "Wir versuchen, die phylogenetischen Beziehungen von 1,8 Millionen Arten herauszufinden, und wir können nicht selbst zwanzig Arten von Hefen sortieren", sagt er.
Um das Paradoxon zu lösen, entwickelten die Forscher einen Algorithmus, der auf der Informationstheorie basiert, um das Vertrauen in die Richtigkeit einzelner Teile des Baums zu messen. Sie hoffen, dass der neue Ansatz dazu beitragen wird, die Evolutionsperioden zu klären, die sowohl die interessantesten und nützlichsten als auch die widersprüchlichsten Daten enthalten - zum Beispiel die
kambrische Explosion - die rasche Diversifizierung des Tierlebens vor 540 Millionen Jahren.
„Historisch gesehen beziehen sich die interessantesten Episoden auf Bereiche, die Aufmerksamkeit erregt und Kontroversen ausgelöst haben“, wie beispielsweise die Herkunft von Tieren, Wirbeltieren und Blütenpflanzen, sagt
Antonis Rokas , Biologe an der Vanderbilt University, der die neue Studie leitete.
Basierend auf den Ergebnissen des neuen Algorithmus können Wissenschaftler nur die informativsten Gene für die Erstellung phylogenetischer Bäume auswählen. Ein solcher Ansatz kann den Prozess sowohl genauer als auch effizienter machen. "Ich denke, es wird helfen, den Wiederaufbau des Baumes des Lebens zu beschleunigen", sagte Khidir Hilu, Biologe am Virginia Institute of Technology.
Ziegel des Lebens
Die Grundlage für phylogenetische Bäume wird durch die Gruppierung von Arten nach ihrem Verwandtschaftsgrad geschaffen. Wenn wir die DNA von Menschen, Schimpansen und Fischen vergleichen, wird klar, dass Menschen und Schimpansen näher beieinander sind als Fische.
Es war einmal, als Forscher ein oder mehrere Gene verwendeten, um Organismen zu vergleichen. In den letzten zehn Jahren gab es jedoch eine Explosion phylogenetischer Daten, die sehr schnell die für die Erstellung dieser Bäume erforderlichen Grundlagen füllten. Die Analyse füllte mehrere der weißen Flecken, die über den Baum verstreut waren, aber es bestehen immer noch ernsthafte Meinungsverschiedenheiten.
Zum Beispiel ist noch nicht klar, wer den Schnecken am nächsten kommt - Muscheln oder Mollusken mit
Schaufelbeinen , sagt Rokas. Es ist nicht genau bekannt, wie einige der frühesten Zweige von Tieren eines Baumes, wie Quallen und Schwämme, miteinander verbunden sind. Wissenschaftler können Beispiele für widersprüchliche Bäume zeigen, die in denselben wissenschaftlichen Zeitschriften mit einem Unterschied von Wochen oder
sogar in derselben Ausgabe erscheinen .
"Daher die Frage: Warum fällt es uns so schwer, uns zu einigen?" - sagt Rokas.

Rokas und sein Doktorand Leonidas Salichos untersuchten dieses Problem, indem
sie Gene einzeln bewerteten und die nützlichsten Gene - mit den meisten Informationen zur Evolutionsgeschichte - verwendeten, um ihre Version des Baumes zu erstellen.
Sie begannen mit 23 Hefearten und wählten 1.070 Gene aus. Zunächst erstellten sie auf übliche Weise einen phylogenetischen Baum, die Verkettung. Dazu werden alle Sequenzen einzelner Arten zu einem Megagen zusammengefasst und anschließend Sequenzen einzelner Arten mit dieser langen Sequenz verglichen, auf deren Grundlage ein Baum erstellt wird, der die Unterschiede am besten erklärt.
Der resultierende Baum ist hinsichtlich der statistischen Standardanalyse genau. Da ähnliche Methoden dazu führen, dass Bäume voller Meinungsverschiedenheiten sind, beschlossen Rokas und Salichos, sich mit dem Thema zu befassen. Sie bauten Sätze phylogenetischer Bäume für einzelne Hefegene und verwendeten einen Algorithmus, der unter Verwendung der Informationstheorie entwickelt wurde, um nach Bereichen mit der größten Übereinstimmung zwischen verschiedenen Bäumen zu suchen. Das Ergebnis,
das im Mai in der Zeitschrift Nature veröffentlicht wurde , war unerwartet. Jedes untersuchte Gen scheint eine etwas andere Evolutionsgeschichte zu erzählen.
„Fast alle Bäume, die für einzelne Gene gebaut wurden, stießen aufgrund der Datenverkettung auf einen Baum“, sagt Hilu. "Das ist schockierend."
Sie kamen zu dem Schluss, dass mehrere Gene, die eine bestimmte Architektur unterstützen, genau sein müssen. Wenn jedoch verschiedene Gengruppen zwei unterschiedliche Architekturen gleichermaßen unterstützen, verringert sich die Wahrscheinlichkeit, dass sie genau mit der Realität übereinstimmen. Rokas und Salichos verwendeten eine Methode namens
statistischer Bootstrap , um die informativsten Gene auszuwählen.
"Wenn Sie nur Gene mit aktiver Unterstützung nehmen, erhalten Sie den richtigen Baum", sagt Donogue.
Der überarbeitete Baum fiel mit einem Baum zusammen, der auf einer alternativen Quelle evolutionärer Informationen aufgebaut war - groß angelegte Veränderungen der von Generation zu Generation übertragenen DNA-Segmente -, die ihre Forschung rechtfertigten.
Entdeckungen waren nicht auf Hefe beschränkt. Bei Anwendung derselben Analyse auf größere und komplexere Lebensformen, einschließlich der genetischen Daten von Wirbeltieren und Tieren, fanden sie schwerwiegende Konflikte zwischen einzelnen Genen.
Einige Forscher müssen sich an die Idee gewöhnen, Daten selektiv von der Analyse auszuschließen. „Seit vielen Jahren besteht das Hauptproblem für Menschen, die versuchen, die Beziehungen von Organismen zu verstehen, darin, genügend Daten zu sammeln“, sagt
Jeffrey Townsend , ein Evolutionsbiologe in Yale, der nicht mit Forschung zu tun hat. "Der Community wurde immer über die Notwendigkeit eines Datensatzes berichtet, daher ist es nicht verwunderlich, dass sie sich der Aufgabe auf diese Weise näherten."
Obwohl Evolutionsbiologen seit Jahren mit diesen Problemen zu kämpfen haben, ist die neue Studie der bislang größte Versuch, das Ausmaß des Konflikts einzelner Gene zu untersuchen. „Die Menschen werden zwei Reaktionen haben: Es gibt mehr Konflikte als ich dachte, und wir müssen lernen, sie besser zu analysieren“, sagt Donague, der die neue Methode in seiner Arbeit anwenden möchte. Er weist jedoch auch auf Schwierigkeiten bei der Bestätigung der Richtigkeit des neuen Ansatzes hin. Obwohl der überarbeitete Baum mit dem übereinstimmt, was auf alternativen genetischen Informationen basiert, kann letztere seine eigenen Inkonsistenzen aufdecken. "Ich bin nicht sicher, ob wir wissen, wie die Beziehung wirklich ist", sagt er. "Und wenn wir uns nicht sicher sind, wie es wirklich läuft, wissen wir nicht, ob wir den richtigen Baum haben."
Bild ändern
Die Forscher müssen die neue Technik breiter anwenden, um zu sehen, wie sie das Konzept der Evolution verändern kann. Rokas und Salichos haben jedoch bereits gezeigt, dass es am schwierigsten ist, kurze Äste des Baumes oder „buschige“ Teile davon zu rekonstruieren, die Perioden schneller Speziation darstellen - insbesondere solche, die näher an der Basis des Baumes und tief in der Evolutionsgeschichte liegen.
"Theoretische Untersuchungen haben dieses Verhalten vorhergesagt, aber unsere Studie zeigt zum ersten Mal eine Bestätigung anhand experimenteller Daten", sagte Rokas.
Rokas argumentiert, dass neue Entdeckungen die Art und Weise verändern werden, wie Forscher unklar gerahmte Teile eines Baumes interpretieren. „Evolutionsbiologen gehen normalerweise davon aus, dass der Baum falsch ist, wenn er nicht über die erforderlichen Details verfügt. Wenn wir also mehr Daten sammeln und bessere Algorithmen erstellen, kommen wir zum richtigen Baum “, sagt er. Das Vorhandensein widersprüchlicher Teile des Baums, die trotz des Datenflusses und der Anwendung einer neuen Art der Analyse bestehen bleiben, kann jedoch auf das Vorhandensein buschiger Teile hinweisen. „Ich denke, in einigen Fällen kann der Algorithmus diesen Konflikt lösen, in anderen ist es möglich, Konfliktbereiche zu markieren, die wir wahrscheinlich nicht lösen können.
Die Untersuchung dieser buschigen Teile des Baumes kann einen neuen Blick auf die besonders interessanten Stadien der Evolution werfen, zum Beispiel die kambrische Explosion, als das Leben von der Vorherrschaft einfacher Organismen auf eine Vielzahl von Tierarten überging.
Andere Wissenschaftler sind sich einig, dass Entdeckungen Einfluss darauf haben können, wie Spezialisten mit widersprüchlichen Vorstellungen über die Evolution umgehen. "Ich denke, dies ist ein Vorbote eines Paradigmenwechsels", sagte Townsend. "Wenn wir geeignete Methoden anwenden, haben wir die Möglichkeit, mehr über Probleme zu erfahren, die uns seit langem plagen."
Townsend, der seine eigene Methode zur Auswahl der informativsten Gene basierend auf
ihrer Evolutionsrate entwickelt hat , stellt fest, dass sich nicht alle Mitglieder der wissenschaftlichen Gemeinschaft über die Notwendigkeit neuer Ansätze einig sind. "Ich hoffe, diese Arbeit hilft, dieses Problem in den Vordergrund zu rücken", sagte er.
Die Auswahl der richtigen Menge an Genen für den Bau von Prototypen phylogenetischer Bäume ist nicht die einzige Frage, die Evolutionsbiologen plagt. Sie müssen sich auch darauf einigen, wie viele Arten in die Verarbeitung einbezogen werden sollen - je mehr Arten im Baum enthalten sind, desto schwieriger ist die Analyse. Die Ergebnisse können auch aufgrund von Unterschieden in der Qualität der für verschiedene Arten gesammelten Daten variieren. „Wenn wir eine echte Evolutionsgeschichte darüber brauchen, wie alles miteinander verbunden ist, was ist dann besser dafür - mehr Gene oder mehr Arten zu sammeln? - sagt Donogue. "Ich denke beides."
Neue Ansätze, mit denen Forscher mit weniger Genen genaue Ergebnisse erzielen können, können den Evolutionsbaum erweitern. Die Möglichkeit, nur das informativste der Gene auszuwählen, kann den Prozess effizienter gestalten und es Wissenschaftlern ermöglichen, mit weniger Daten und Ressourcen genaue Bäume zu erstellen. „Wenn wir mehrere Gene auswählen und den gleichen guten Baum wie das gesamte Genom erhalten könnten“, sagt Khilu, „könnten wir einen viel detaillierteren Lebensbaum erstellen - auf der Ebene der Gattungen oder sogar auf der Ebene der Arten - anstatt uns damit zufrieden zu geben das Skelett der wichtigsten Zweige. "