Die Analyse der Zugehörigkeit zur menschlichen Bevölkerung durch DNA wirft nach unserer Erfahrung drei große Fragen in der Öffentlichkeit auf: Können Gene und ethnische Gruppen miteinander verknüpft werden, wie erfolgt die Analyse der Herkunft aus technischer Sicht und ob Gentests „Juden identifizieren“ können? Aus irgendeinem Grund ist es genau das Problem der jüdischen Identität mit der DNA, das sowohl diejenigen beunruhigt, die unbestreitbare Beweise für die Zugehörigkeit zu dem von Gott auserwählten Volk haben, als auch diejenigen, die keine Matze essen und die Thora nicht lesen.
Im neuen Genotek-Material zu Geektimes werden wir versuchen, alles in der richtigen Reihenfolge zu beantworten. Und ja, wir definieren auch Juden.

Rennen alias Bevölkerungsgruppen in Biologie, Medizin und Genetik
Die Menschheit hat die schlechte Angewohnheit, Gewalt mit der „angeborenen“ Überlegenheit einer Rasse gegenüber einer anderen zu rechtfertigen - deshalb gehen moderne Biologen das Problem der genetischen Unterschiede zwischen Populationen mit größerer Vorsicht an. Die (Nicht-) Existenz biologischer Grenzen zwischen Rassen und ethnischen Gruppen wurde im gesamten 20. Jahrhundert vehement diskutiert, es wurde jedoch noch kein endgültiger Konsens zu diesem Thema erzielt (
1 ).
Es wurde gehofft, dass die Sequenzierung des menschlichen Genoms alle zusammenbringen würde. Das Genom, gelesen von „von“ und „bis“, zeigt, dass die Grenzen zwischen Gruppen sozialer Natur sind und die Gene für alle gleich sind. Es stellte sich anders heraus: Eine sorgfältige Untersuchung des menschlichen Nukleotidcodes belebte und erhöhte das Interesse an den biologischen Unterschieden zwischen rassischen und ethnischen Bevölkerungsgruppen. Dieselben Gene fanden im Allgemeinen leicht unterschiedliche Allelvarianten, die mit dem Risiko von Krankheiten (
2 ), dem Arzneimittelstoffwechsel (
3 ) und der Reaktion des Körpers auf Umweltbedingungen (
4 ) verbunden waren, und diese Varianten wurden in verschiedenen Populationen mit unterschiedlichen Häufigkeiten gefunden.
Die Suche nach nicht existierenden "indischen" oder "afrikanischen" Genen wurde eingestellt, aber die Forschung auf dem Gebiet der Medizin- und Populationsgenetik zieht immer noch Parallelen zwischen den biologischen Merkmalen und der ethnischen Zugehörigkeit der Teilnehmer. Die Verwendung der Begriffe „Rasse“ und „ethnische Zugehörigkeit“ in solchen Werken wird aktiv diskutiert (und oft verurteilt). Es gab Versuche, Regeln einzuführen, die die Forscher dazu zwingen, die Notwendigkeit der Verwendung von „rutschigen“ Kategorien zu rechtfertigen und zu klären, was genau unter bestimmten Begriffen zu verstehen ist. Im Februar letzten Jahres veröffentlichte Science, eine der angesehensten Fachzeitschriften, einen mehrdeutigen Artikel (
5 ), in dem vorgeschlagen wurde, die Verwendung des Begriffs „Rasse“ in der Genforschung vollständig aufzugeben und ihn durch eine korrektere und neutralere „Abstammung“ - „Herkunft“ - zu ersetzen. .
Aber auch unter unsicheren Bedingungen ist die Menschheit immer noch in Bevölkerungsgruppen unterteilt: insbesondere für die korrekte Durchführung klinischer Arzneimittelstudien und die Bewertung des Krankheitsrisikos. Beispielsweise sind drei allelische Varianten des NOD2-Gens - R702W, G908R und 1007fs - mit einem erhöhten Risiko für Morbus Crohn bei Europäern verbunden (
6 ,
7 ), jedoch ist keine dieser Varianten mit Morbus Crohn bei Japanern assoziiert (
8 ). Es ist bekannt, dass Allele des CCR5-Gens die Entwicklungsrate von Immundefekten bei HIV-infizierten Patienten beeinflussen (
9 ): Unter ihnen wurde eine Option gefunden, die das Fortschreiten der Krankheit bei Amerikanern europäischer Abstammung verlangsamt, aber ihre Entwicklung bei Afroamerikanern beschleunigt (
10 ). Asiaten fanden eine Korrelation zwischen Polymorphismen des p53-Proteingens, das die Stressreaktion reguliert und die Entwicklung von Tumoren unterdrückt, und durchschnittlichen Wintertemperaturen in den Lebensräumen von Populationen - genetische Anpassung an Frost (
11 ). Und wenn in der Vergangenheit nur Informationen verwendet wurden, die von den Teilnehmern selbst bereitgestellt wurden, um die Stichprobe in ethnische Gruppen aufzuteilen, werden sie in der postgenomischen Ära zunehmend durch eine genetische Bewertung der Herkunft des Subjekts ergänzt und verfeinert.
Genetische Variation zwischen Populationen
Im Alltag teilen wir Menschen nach Aussehen oder Kommunikationssprache in Gruppen ein. Die meisten Dänen ähneln sich mehr als jeder Italiener (
hier eine coole Visualisierung mit gemittelten Porträts verschiedener Nationalitäten). Die Dänen und Italiener sind einander viel näher als jeder von ihnen - den Bewohnern Afrikas südlich der Sahara: Die Phänotypen des Menschen sind nach geografischen Mustern gruppiert. Die Verteilung der Genotypen hat eine ähnliche Struktur: Mitglieder einer lokalen Gruppe haben in der Regel engere familiäre Bindungen als Bewohner abgelegener Gebiete, und die in einer Region lebenden Bevölkerungsgruppen sind näher als diejenigen, deren Lebensräume durch geografische Barrieren (z. B. Gebirgszüge oder Wasser) getrennt sind Array).
Darüber hinaus ist die genetische Vielfalt der menschlichen Bevölkerung geringer als die vieler biologischer Arten. Dies erklärt sich aus der Tatsache, dass die Menschheit eine junge Spezies ist: Einzelne Gruppen hatten relativ wenig Zeit, um Unterschiede zu akkumulieren. Zwei zufällig ausgewählte Personen unterscheiden sich durch jeweils ~ 1000 Nukleotide voneinander, während die beiden Schimpansen in ~ 500 "Buchstaben" nicht einmal zusammenfallen. Insgesamt gibt es jedoch rund 3 Millionen potenzielle „Unterschiede“ im menschlichen Genom. Die meisten dieser Diskrepanzen, die als Einzelnukleotidpolymorphismen (SNPs) bezeichnet werden, sind neutral oder nahezu neutral, aber einige von ihnen sind für phänotypische Unterschiede zwischen Menschen verantwortlich.
Die Verteilung neutraler Polymorphismen (da sie keine biologische Bedeutung haben, keiner gerichteten evolutionären Selektion unterliegen, vom Wind der Migrationen getragen werden) in der Weltbevölkerung spiegelt die demografische Geschichte unserer Spezies wider. Genetische und archäologische Beweise weisen darauf hin, dass die Größe der menschlichen Bevölkerung in den letzten 100.000 Jahren erheblich zugenommen hat. Menschen ließen sich außerhalb Afrikas nieder und kolonisierten den Rest der Welt. Der Neuansiedlungsprozess wirkte sich auf zweierlei Weise auf die geografische Verteilung der Allele aus: Erstens war der „Gründereffekt“ betroffen - in der Einwandererbevölkerung war in der Regel nur ein Teil der genetischen Varianten aus dem gesamten Pool ihrer Vielfalt in der Ahnenpopulation vertreten; zweitens fand die sogenannte "assortative Kreuzung" statt, d.h. Paare bildeten sich hauptsächlich innerhalb ihrer Gruppe, was die Verteilung bestehender und aufkommender De-novo-Polymorphismen unter Individuen, die in verschiedenen geografischen Gebieten leben, einschränkte. Diese Prozesse führten zu einer allmählichen Anhäufung genetischer Unterschiede.
Im Kontext von Bevölkerungsgruppen wurden in den 70er bis 80er Jahren genomische Marker untersucht, in den 90er Jahren wurden sie verwendet, um die Bevölkerung einer bestimmten Person zu identifizieren. Forscher haben wiederholt gezeigt, dass genetische Polymorphismen Bevölkerungsgruppen erfolgreich isolieren und die Gruppenzugehörigkeit eines Individuums bestimmen können. Dann wurde gezeigt, dass Menschen, die auf demselben Kontinent leben, normalerweise genetisch näher beieinander sind als Menschen aus verschiedenen Kontinenten. In solchen Studien waren Informationen über Geburtsort, Rasse und ethnische Gruppe von Anfang an bekannt und wurden in Verbindung mit genetischen Daten verwendet. Wenn die Probanden nur aufgrund genetischer Merkmale blind auf die Cluster verteilt wurden, war die Entsprechung zwischen geografischer Herkunft, ethnischer Zugehörigkeit und Bevölkerungsstruktur weniger ausgeprägt. Wie weitere Studien gezeigt haben, hing der Erfolg von den verwendeten genetischen Markern und ihrer Anzahl (mehr ist besser), der richtigen Auswahl der Referenzpopulationen und anderen Faktoren ab (
12 ).
In den USA wurde die genetische Definition der Bevölkerung bis 2004 nicht nur in der biomedizinischen Forschung, sondern auch bei Kriminaluntersuchungen verwendet:
Dieser Artikel von Nature enthält eine spannende Geschichte darüber, wie Polizisten, die verzweifelt nach einem Verbrecher suchen, einen DNA-Test bei einem Handelsunternehmen angeordnet haben Hautfarbe des Verdächtigen und öffnete den Fall. Vorschläge zur Analyse der genetischen Herkunft haben die Welle des allgemeinen Interesses an Menschen in ihrer eigenen Vergangenheit erfolgreich getroffen. "Roots Mania", so genanntes Hobby, in einem Artikel in Time, der sich "Amerikas jüngster Besessenheit" widmet - genealogische Forschung.
Genomische Methoden werden von Spezialisten aktiv eingesetzt, die den Ursprung und die Entwicklung von Menschen untersuchen. Beispielsweise hat ein internationales Forscherteam 2013 mithilfe einer genetischen Analyse die Hypothese der Herkunft aschkenasischer Juden aus den Khazaren widerlegt (
13 ). Der von den Autoren verwendete Genomdatensatz ist gemeinfrei: Er enthält mehr als 100 Weltpopulationen. Wir schlagen vor, mit uns eine kleine Studie zu simulieren: den Ort der Genotek-Kunden in dieser Stichprobe zu bestimmen und gleichzeitig die technischen Details der Bevölkerungsbestimmung zu verstehen.
Forschungszweck
Identifizieren Sie Genotek-Kunden unter Referenzpopulationen. Finden Sie heraus, ob sich in unserer Stichprobe Vertreter aschkenasischer Juden befinden. Demonstrieren Sie die Prinzipien und Methoden der Analyse der Bevölkerung eines Individuums.
Forschungsziele
Verarbeiten Sie die Genotypisierungsdaten von 722 Probanden mit dem ADMIXTURE-Programm unter Verwendung des Datensatzes von Behar et al., 2013 als Trainingsprobe.
Materialien und Methoden
Die ursprüngliche Arbeit von Behar et al., 2013, verwendete Daten von 1.774 Personen: Unter ihnen befanden sich Vertreter von 88 nichtjüdischen Bevölkerungsgruppen (aus Arabien, Zentralasien, Ostasien, Europa, dem Nahen Osten, Nordafrika, Sibirien, Südasien und Subasien) Sahara-Afrika) und 18 jüdische Bevölkerungsgruppen. Die Autoren benötigten einen umfangreichen Datensatz, um den Ort der Aschkenasen im Kontext der Weltbevölkerung genau zu bestimmen: Die Aufgabe bestand darin, alle drei geografischen Regionen darzustellen, aus denen diese Gruppe hypothetisch stammen könnte - Europa, den Nahen Osten und das Khazar Khaganate. Die Autoren betonten den Unterschied zwischen dem Ansatz der Probenahme, der moderne europäische, nahöstliche und jüdische Bevölkerungsgruppen repräsentiert - direkte Nachkommen von Ahnenpopulationen - und Stichproben, die dem Khazar Kaganate entsprechen, das vor etwa 1000 Jahren nicht mehr existierte. Der Haken ist, dass keine der existierenden Bevölkerungsgruppen der direkte Erbe des Khaganats ist. Als mögliche moderne Vertreter der Khazaren wählten die Autoren die Bewohner des Südkaukasus (Abchasen, Armenier, Aserbaidschaner, Georgier), des Nordkaukasus (Adygs, Balkare, Tschetschenen, Kabarden, Osseten und verschiedene andere Nationalitäten), Tschuwaschien und Tataren.
Wir haben dem Datensatz Stichproben von 722 Personen aus verschiedenen Regionen Russlands hinzugefügt.
Für die statistische Analyse verwendeten wir das Programm ADMIXTURE, mit dem wir den wahrscheinlichsten Ursprung eines Individuums anhand von Daten zu Genotypen abschätzen können. Darüber hinaus verwendeten die Autoren des diskutierten Artikels andere statistische Methoden, die eine ähnliche Antwort auf die gestellte Frage gaben. Wir werden uns auf ADMIXTURE konzentrieren, da dieser Algorithmus es uns ermöglicht, den prozentualen Beitrag der Ahnenpopulationen zu den untersuchten Genomen abzuschätzen.
ADMIXTURE verwendet Monte-Carlo-Methoden in Markov-Ketten (Markov-Kette Monte Carlo, MCMC). Hier ist ein
Link zu einem Artikel der Autoren des Algorithmus für diejenigen, die die mathematische Seite des Prozesses genauer verstehen möchten.
Mal sehen, wie ADMIXTURE am Beispiel von Proben und Populationen aus unserem Set funktioniert
Insgesamt haben wir 2.496 Proben / Individuen, von denen jede zu einer von 106 modernen Populationen gehört. Wir schlagen vor, dass moderne Populationen höchstwahrscheinlich von einer relativ kleinen Anzahl von Ahnenpopulationen abstammen. Die "Ahnenpopulationen" in dieser Analyse sind einige alte Genomcluster, die durch das Prinzip der genetischen Ähnlichkeit vereint sind. Mit ADMIXTURE können sowohl willkürlich Annahmen über die Anzahl solcher Cluster in der Stichprobe getroffen als auch die optimale Anzahl ausgewählt werden, die die tatsächliche Verteilung der Genomdaten am korrektesten beschreibt.
Nachdem ADMIXTURE Informationen über die Genotypen und die geschätzte Anzahl der "Ahnen" -Populationen (K) erhalten hat, erstellt es ein Modell, das den Beitrag jeder der "Ahnen" -Populationen zu jeder Stichprobe schätzt. Bei der Interpretation der Daten sind sowohl die quantitative Zusammensetzung des Genoms (Prozentsatz der Cluster) als auch die qualitative wichtig - ihre Anwesenheit oder Abwesenheit in bestimmten Genomen. Basierend auf diesen Daten kann man Annahmen über Evolutionsprozesse in einer Population treffen, insbesondere über das Vorhandensein oder Fehlen gemeinsamer „Wurzeln“ in Bevölkerungsgruppen. Die Schlussfolgerungen sind jedoch legitim, wenn das von uns konstruierte Modell gut ist: Der optimale Wert von K. wird ausgewählt.
Wir wählen den optimalen Wert von K.
Wie kann festgestellt werden, wie viele „Ahnen“ -Populationen für eine bestimmte Stichprobe am ehesten mit der wahren übereinstimmen? Empirisch!
ADMIXTURE ist ein intelligentes Programm: Sie erstellt ein Modell der genetischen Struktur von Populationen auf der Grundlage von Daten zu einzelnen Genotypen (Bewertung des Beitrags jedes der alten Genomcluster zu jedem der Probengenome) für eine bestimmte Zahl K und vergisst nicht, am Ende einen Vergleich mit der Realität anzustellen. Überprüfen Sie, wie gut die Eingabe durch das konstruierte Modell beschrieben wird. Ein Vergleichsmaß ist der „Fehler“ - ein Wert, der die Nichtübereinstimmung zwischen dem Modell und den realen Daten beschreibt. Je größer der Fehler ist, desto schlechter ist die Annahme, dass die Anzahl der Ahnenpopulationen wahr ist.
Wie wählt man den optimalen Wert von K? Wir starten den ADMIXTURE-Algorithmus für dieses Beispiel, indem wir verschiedene Werte von K ersetzen, und wir erhalten für jedes K seinen eigenen Fehlerwert. Wir zeichnen die Abhängigkeit der Größe des Fehlers von K auf. Hier ist die Grafik, die von den Autoren des Artikels erhalten wurde:
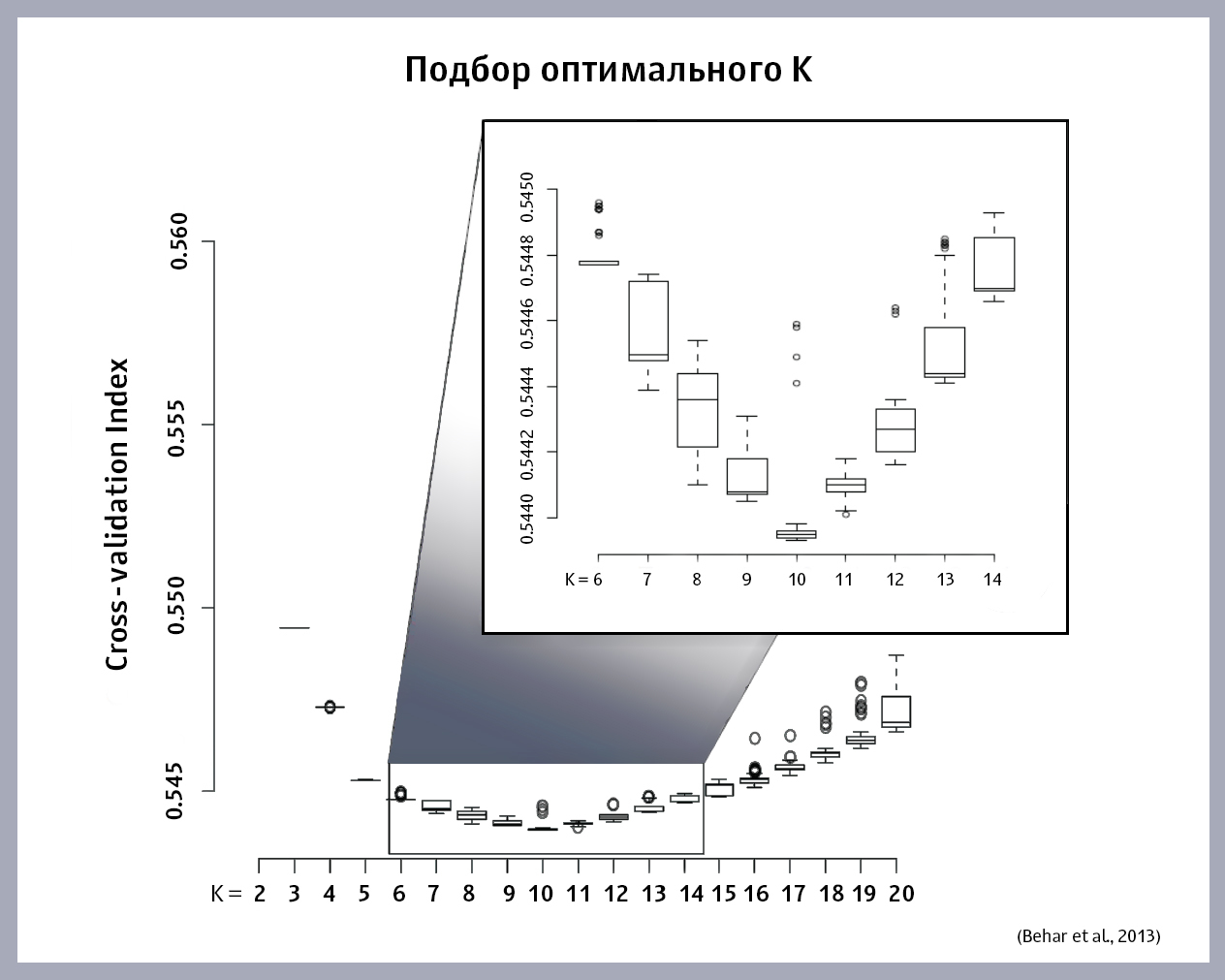
Der optimale Wert von K liegt am Minimalpunkt der Funktion. Wenn das Minimum im Diagramm nicht gefunden wird (die Funktion wächst oder nimmt ständig ab), müssen Sie Modelle erstellen, indem Sie neue Ks auswählen, bis Sie das richtige finden.
Selbst bei optimal ausgewähltem K hängt die Zuverlässigkeit der Analyseergebnisse von der Richtigkeit der Probe ab:
1. Einzelpersonen sollten nicht miteinander verwandt sein.
2. Einzelnukleotidpolymorphismen (SNPs), die zur Genotypisierung verwendet werden, sollten mit einer ausreichend hohen Dichte gleichmäßig über das Genom verteilt sein.
3. SNP-Allele müssen sich im Gleichgewicht befinden, dh die Wahrscheinlichkeit des Vorhandenseins eines bestimmten Allels in einem bestimmten Individuum sollte nur von der Häufigkeit dieses Allels in der Population und nicht von anderen Allelen im Genom abhängen.
Wie aus der Grafik ersichtlich ist, betrug das optimale K für diese Stichprobe 10 "Ahnen" -Populationen.
Ergebnisse
Die Analyseergebnisse werden von ADMIXTURE folgendermaßen visualisiert (in der Abbildung ist nur ein Teil der Daten sichtbar):
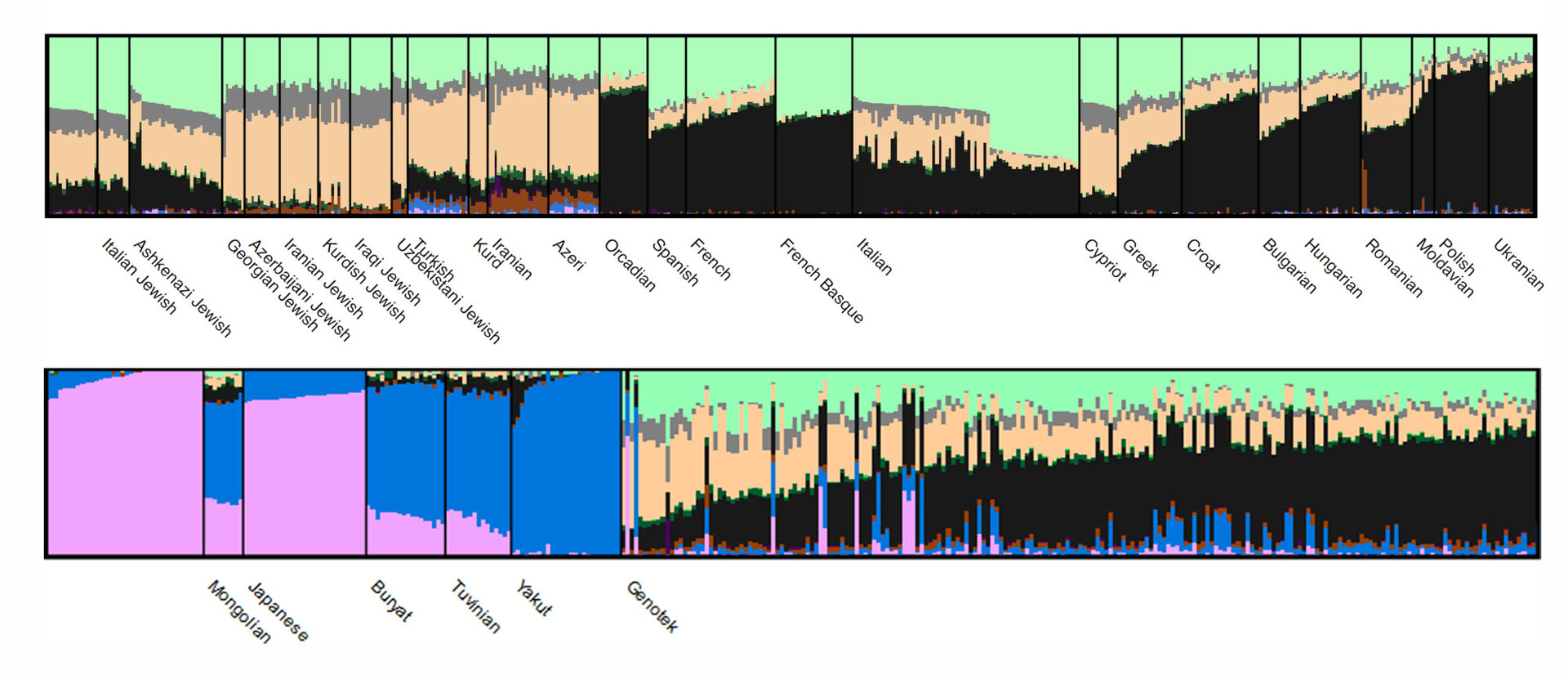
Jeder Cluster hat seine eigene Farbe, und die Populationen unterscheiden sich (oder unterscheiden sich nicht) in den Anteilen der Cluster im Genom.
Hier liegt die interaktive Version des Bildes für eine detaillierte Studie: Bewegen Sie die Maus und scrollen Sie, um alle Populationen zu sehen oder einige der Gruppen genauer zu betrachten.
Im Allgemeinen wird erwartet, dass innerhalb der Genotek-Population das Cluster-Verhältnis dem Muster entspricht, das für Populationen osteuropäischer Herkunft charakteristisch ist. Das Interessante beginnt auf der Ebene der einzelnen Proben:
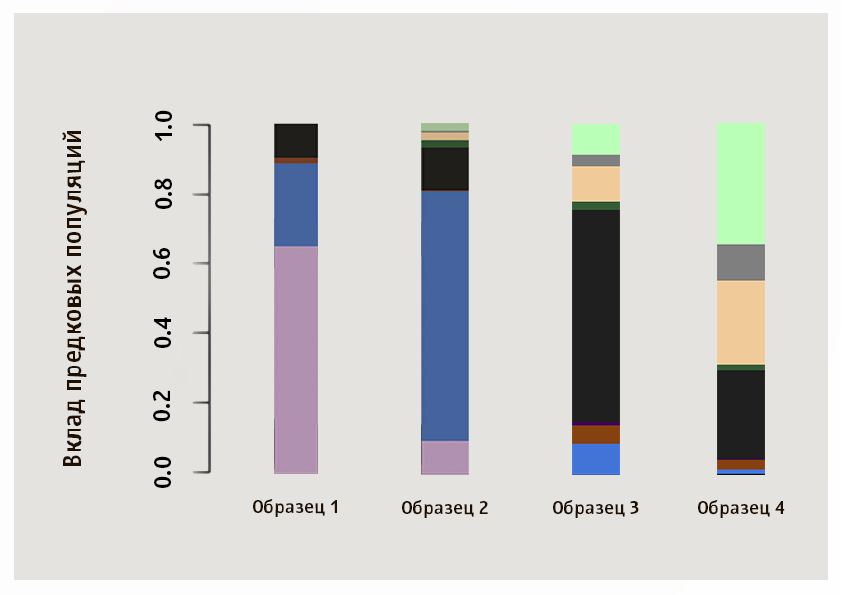
Obwohl die Population, die der gegebenen Stichprobe am nächsten liegt, durch numerische Werte bestimmt wird, können viele Informationen auch durch visuellen Vergleich von Mustern erhalten werden. Wir empfehlen, dass Sie unabhängig voneinander die nächstgelegenen Populationen für Stichproben von vier Genotek-Kunden aus dem Bild bestimmen.
Die AntwortIn diesem Bild sind die Proben 1 und 2 asiatischen Ursprungs: Die Dominanz des rosa Clusters ist typisch für die Japaner und die Khan in unserer Stichprobe, blau für die Jakuten, die dritte Stichprobe zeigt das für Russen, Weißrussen, Ukrainer und Polen typische Verhältnis der Komponenten und die vierte ist typisch Ashkenaz Jude.
Aus 722 Proben fanden wir 9 aschkenasische Juden.
Fazit
Die Zugehörigkeit zur Bevölkerung ist bei weitem nicht der einzige Faktor, der die ethnische Selbstidentifikation einer Person bestimmt. Es ist jedoch weiterhin möglich, eine Korrelation zwischen ethnischen Gruppen und der Struktur des Genoms ihrer Vertreter aufzudecken. Eine solche Analyse wird sowohl für wissenschaftliche und medizinische Zwecke als auch zur Untersuchung ihrer eigenen Wurzeln durch alle Ankömmlinge verwendet. Gleichzeitig ist es wichtig zu verstehen, dass die Modelle ständig verbessert werden, und die Ergebnisse, die für eine höhere Genauigkeit erzielt werden, sollten in Verbindung mit anderen Daten, beispielsweise dem Stammbaum der Familie, berücksichtigt werden.
Von den Autoren des Originalartikels wurden keine Beweise für den khazarischen Ursprung von Ashkenazi gefunden. Gentests „wissen“ natürlich, wie man Juden identifiziert - man sollte jedoch nicht vergessen, dass „Judentum“ in erster Linie ein Geisteszustand ist.
In naher Zukunft wird bei Genotek der aktualisierte Genealogie-DNA-Test mit erweiterten Ergebnissen eingeführt: Wir werden die Anzahl der Populationen auf Hunderte erhöhen und jüdische Populationen hinzufügen. Wir werden die Informationen in Ihrem persönlichen Konto für alle aktualisieren, die uns jemals ihr genetisches Material übergeben haben. Wenn Sie noch nicht genotypisiert sind, laden wir Sie ein, sich anzumelden .Referenzliste
- Foster M., Sharp R. (2002). Rasse, Ethnizität und Genomik: Soziale Klassifikationen als Proxies biologischer Heterogenität. Genom Res.
- Collins FS, McKusick VA (2001). Implikationen des Humangenomprojekts für die Medizin. JAMA.
- Nebert DW, Menon AG (2001) Gene für Pharmakogenomik, Ethnizität und Suszeptibilität. Pharmakogenomik J.
- Olden K., Guthrie J. (2001). Genomik: Implikationen für die Toxikologie. Mutat. Res.
- Yudell M., Roberts D., DeSalle R., Tishkoff S.(2016). Taking race out of human genetics. Science.
- Ogura, Y. et al. (2001). A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature.
- Hugot, JP et al. (2001). Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature.
- Inoue, N. (2002). Lack of common NOD2 variants in Japanese patients with Crohn's disease. Gastroenterology.
- Martin, MP et al.(1998). Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science.
- Gonzalez, E. et al.(1999). Race-specific HIV-1 disease-modifying effects associated with CCR5 haplotypes. Proc. Natl Acad. Sci. USA
- Shi, Hong et al. (2009). Winter Temperature and UV Are Tightly Linked to Genetic Changes in the p53 Tumor Suppressor Pathway in Eastern Asia. American Journal of Human Genetics.
- Bamshad M., Wooding S., Salisbury B. et al. (2004). Dekonstruktion der Beziehung zwischen Genetik und Rasse. Nat Rev Genet.
- Behar DM et al. (2013). Keine Hinweise aus genomweiten Daten khazarischen Ursprungs für die aschkenasischen Juden. Humanbiologie