 Sie können so viel sagen, wie Sie möchten, dass in Russland etwas mit der Medizin nicht stimmt, aber unser Verbraucherschutz ist einer der besten der Welt.
Sie können so viel sagen, wie Sie möchten, dass in Russland etwas mit der Medizin nicht stimmt, aber unser Verbraucherschutz ist einer der besten der Welt. Im Vergleich zu unseren europäischen Standards für Medizinprodukte ist es nur ein Filkin.
In den letzten Jahren müssen wir jeden Effekt nachweisen. Das heißt, wenn Sie auf eine Salbe aus Falten stoßen, die jedoch keine Falten entfernt, können Sie sich bei Rospotrebnadzor beschweren. Und irgendwo weit weg in der Branche hört man das Öffnen eines Glases Vaseline. Wenn Sie sich nach der Einnahme von Heilbalsam krank fühlen, können Sie sich auch beschweren. Und wieder - ein Mann, sie werden mit einem Scheck zu jemandem kommen.
Diese ganze Geschichte der Verschärfung der Anforderungen hat uns vor ein paar Jahren, als wir Blefarogel registrierten, ernsthaft getroffen. Aber lassen Sie mich Ihnen im Detail sagen, wie viel Vaseline, Tränen und Rotz wir für diesen Prozess ausgegeben haben.
4 Kreise der Hölle
Grundlage für die Registrierung von Medizinprodukten ist das
Dekret Nr. 1416 .
Es beschreibt die Regeln und Verfahren für die Registrierung. Um dieses ziemlich komplizierte Verfahren durchführen zu können, müssen die folgenden Tests durchgeführt werden:
- Toxikologisch (Nachweis, dass das Produkt in Bezug auf die in der Formel verwendeten Materialien und Substanzen sicher ist).
- Technisch (Nachweis, dass das Produkt in seinen Eigenschaften und technischen Merkmalen den Anforderungen der TU und der Betriebsdokumentation des Herstellers entspricht sowie dass der Produktionsprozess einheitlich ist und nicht jedes einzelne Element der Charge überprüft werden muss).
- Klinisch (Nachweis der Wirksamkeit und Sicherheit der Lösung eines medizinischen Problems).
- Und eine Prüfung der Qualität, Effizienz und Sicherheit, bei der die Kommission alle Daten zum Produkt auf einem Haufen sammelt und über die Möglichkeit seiner Registrierung und Markteinführung entscheidet.
Ein wichtiger Punkt:
Laut Gesetz kann ein Medizinprodukt nicht heilen. Das heißt, wenn es eine therapeutische Wirkung hat, dann ist dieses Arzneimittel eine völlig andere Kategorie. Das medizinische Gerät kann dabei ein Hilfsmittel sein. Wenn beispielsweise ein Tonometer aufgezeichnet wird, muss nachgewiesen werden, dass es den Druck misst, dass die Hülse an den Berührungspunkten mit der Haut keine Allergien oder verschiedene Dermatitis verursacht und dass der Druck während der gesamten Lebensdauer des Geräts mit einer bestimmten Toleranz gemessen wird. Bei einer Ente muss ihre Hypoallergenität und Sicherheit für den Patienten nachgewiesen werden. Bei unseren Gelen geht es hauptsächlich darum, Roszdravnadzor einschließlich Toxikologie zu durchlaufen.
Die Toxikologie war früher relativ einfach. Heute müssen wir mit der Komponentenanalyse und ihren Interaktionen beginnen. Wichtig ist, dass dies nicht nur das Produkt selbst betrifft, sondern auch die Verpackung. Früher überprüften sie nur das Gel und jetzt nach neuen Standards das Gel und die Verpackung (Flasche mit Deckel) aller verwendeten Verpackungen und Farben sowie deren Wechselwirkung. Dies geschieht so, dass die Substanz der Flasche die Eigenschaften des Gels nicht verändert und in seine Zusammensetzung wandert.
Zuerst müssen Sie eine Literaturrecherche durchführen, dh Informationen in Form von Artikeln und Rezensionen zu registrierten Analoga finden, die ihre Wirksamkeit und Sicherheit nachweisen, die Praxis für Wirkstoffe finden und nachweisen, dass sie in der Mischung sicher sind.
Warum ist die erste Stufe der Literaturrecherche? Ich möchte Sie daran erinnern, dass wir kein Medikament haben, sondern ein Medizinprodukt. Die neue Formel muss nicht getestet werden. Am häufigsten.
Es sieht so aus: Wir verweisen auf Artikel zur Prüfung, das heißt, wir finden Zeitschriften in den Archiven, wir beglaubigen Screenshots und Kopien, damit die Kommission selbst nicht später nachschaut.
Dann brauchen wir Feedback aus der klinischen Praxis zu registrierten Analoga. Das heißt, wir müssen Veröffentlichungen finden (oder das nächste Analogon nehmen, das bereits einmal registriert wurde, und prüfen, wie es dieses Problem löst). Auf der Grundlage der gesammelten Informationen zu den registrierten Analoga wird ein Akt zur Bewertung der Ergebnisse klinischer Studien mit Medizinprodukten erstellt.
Als nächstes werden wir mit einem Paket von Dokumenten, die den Abschluss aller oben angegebenen Schritte bestätigen, sowie mit anderen zusätzlichen Dokumenten für das Medizinprodukt zur Registrierung nach Roszdravnadzor geschickt.
Wie läuft der Prozess ab?
Zunächst werden Roszdravnadzor ein Antrag und ein Dossier für das zukünftige Produkt vorgelegt. Die Anwendung beschreibt den Zweck, die Risikoklasse, den Umfang und andere Informationen zum Medizinprodukt sowie Informationen zum Hersteller. Zusätzlich zum Antrag ist eine Reihe von Dokumenten beigefügt: Protokolle, Zertifikate aller durchgeführten Tests, technische Dokumentation (TU), Betriebsdokumentation (Gebrauchsanweisung, Etiketten), fotografische Bilder, ein Risikoanalysebericht, Informationen zu behördlichen Dokumenten, ein Qualifikationstestzertifikat und Mietverträge und andere Herstellerdokumente.
Das heißt, zum Zeitpunkt der Einreichung des Antrags müssen alle internen Tests durchgeführt werden (in unserer Produktion, in unserem eigenen Labor sind sie härter als in staatlichen Laboratorien, da es elementar rentabler ist, beim ersten Mal zu gehen und später im Verkaufsprozess nicht auf Party-Reviews zu stoßen). Dann geben Sie die Toxikologie an eines der Laboratorien mit staatlicher Akkreditierung weiter.
In unserem Fall ist beispielsweise
Blefarogel (ein Mittel zur Augenlidhygiene bei der Vorbeugung
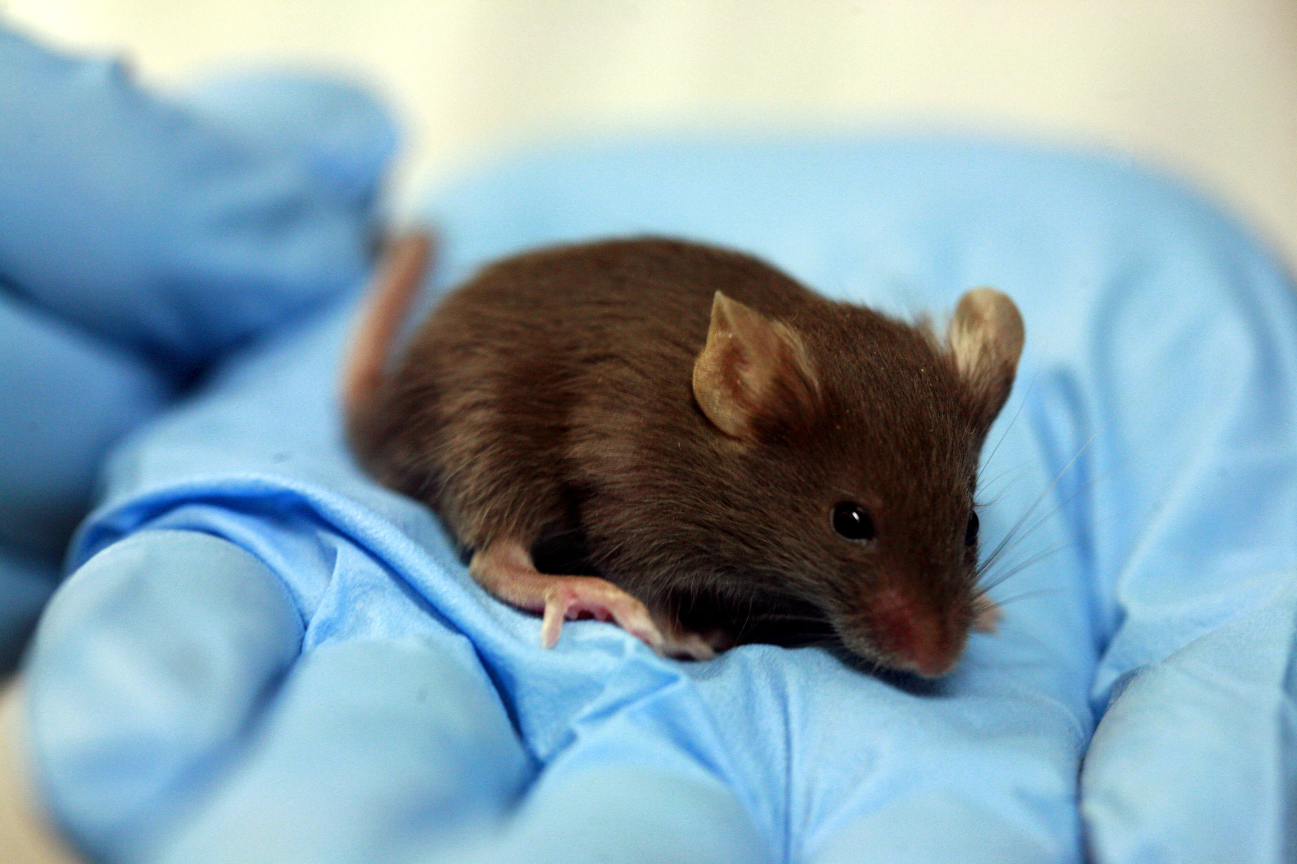
des Syndroms des trockenen Auges ) ein Gel, das auf die Augenlider und die Gesichtshaut aufgetragen wird. Toxikologie waren Mäuse. Ich weiß nicht genau, was sie mit ihnen gemacht haben, aber ich nehme an, dass sie zwei Bereiche an jedem Tier rasiert haben, einen mit Gel verschmiert, den zweiten links oder mit etwas Neutralem verschmiert. Und beobachtete die Maus. Zum Glück schoben sie sich nicht in die Maus. (Okay, sie haben es fast nicht geschoben - Tatsache ist, dass im Sicherheitsdatenblatt [oder Sicherheitsdatenblatt - Sicherheitsdatenblatt] von Propylenglykol Daten zur tödlichen Dosis bei oraler Einnahme enthalten sind. Es benötigt 4 Gramm pro Maus [es bricht höchstwahrscheinlich gleichzeitig physisch] Und schließlich haben sie diese Dosis irgendwie festgelegt ...) Aber kehren wir zu den erfolgreicheren Mausprodukten zurück, die einfach verschmiert sind. Sie haben Nachbarn, die saubere Flaschen aus dem Produkt bekommen haben, in die sie Wasser gießen und verteidigen. Dann werden sie mit diesem Wasser aus den Flaschen verschmiert, um zu überprüfen, ob der Kunststoff im Laufe der Zeit nichts Gefährliches freisetzt. Oder auch nicht: Die genaue Methodik wurde den Herstellern nicht bekannt gegeben. Es besteht eine hohe Wahrscheinlichkeit, dass dieses Wasser aus der Flasche einfach chemisch analysiert und, selbst wenn vermutet wird, intradermal injiziert wird.
Die Mäuse tun uns leid, aber der Ansatz ist folgender: Ohne unsere Tests wären sie überhaupt nicht geboren worden. Im Allgemeinen werden sie dort nicht gequält (zumindest kommen wir bereits mit einer fertigen Studie über uns selbst herein). Wir wissen auch, dass Laborassistenten ein warmes Gefühl für Mäuse haben, vergleichbar mit der Liebe eines Dorfbewohners zu Hühnchen. Und wir vermuten, dass die Maus am Ende der Forschung nicht vollständig entsorgt wird, sondern vor neuen Tests zur Rehabilitation geschickt wird. Dies ist jedoch nur eine Theorie: Wenn hier jemand ist, der über sein Schicksal berichten kann, wäre es wunderbar.
Dem gleichen Antrag muss technische Forschung beigefügt sein. Schreiben Sie insbesondere TU (Zulassungsdokument für ein Medizinprodukt, das technische Anforderungen festlegt) und beweisen Sie, dass das Medizinprodukt diesem entspricht. Experten sind sich über unsere Bedingungen einig. Anschließend werden dort die Stabilität der Serienproben und die Konformität ihrer Produktdossiers überprüft: Farbe, Geruch, elektrische Leitfähigkeit, Dämpfung des Ultraschallsignals usw. - alles, was angezeigt wird. In dieser Phase ist es häufig erforderlich, ein bestimmtes Wort in der Produktbeschreibung neu zu formulieren. Unser Mediagel hat eine andere Viskosität: Dies ist ein Registrierungszertifikat, aber drei Testsätze gelten für mittelviskose, flüssigere und dichtere Gele.
Wir führen in unserer eigenen Produktion die gleichen regelmäßigen Überprüfungen auf Einhaltung der TU durch. Nur einmal in der gesamten Praxis gab es einen Fall, in dem wir uns den zulässigen Grenzen des Tests näherten. Normalerweise gehen wir genau in der Mitte der Toleranzen zwischen den Grenzen.
Die Risikoklasse des Produkts wird durch Zweck, Verwendungsmethode, Verwendungsdauer, Invasivität und andere Kriterien bestimmt. Wir haben Ultraschallgele, zum Beispiel ist die Dauer des Hautkontakts bis zu einer Stunde die erste Klasse. Und in der zweiten Klasse, zum Beispiel bei gynäkologischen Spiegeln, fallen verschiedene Dinge für Neugeborene.
Es gibt einen Unterschied in klinischen Studien. Erstklassige klinische Studien können sofort durchgeführt werden. Die zweite Klasse und höher - die Klinik nur nach der offiziellen Erlaubnis von Roszdravnadzor. Vor klinischen Studien hat das Entwicklungsteam das Recht, nur an sich selbst zu testen. Ziel ist es, den Haupteffekt zu überprüfen. Das Ziel von Beta ist es, mögliche Nebenwirkungen und verschiedene interessante Fälle zu verstehen, z. B. "während der Einnahme anderer Medikamente". Klinische Studien beginnen, wenn die Sicherheit des Medizinprodukts theoretisch nachgewiesen ist.
Ich stelle fest, dass, wenn es nicht um medizinische Produkte, sondern um Kosmetika geht, Studien an Freiwilligen in Kliniken durchgeführt werden, die für wissenschaftliche Forschung dieser Art akkreditiert sind.
In der Praxis sieht dies folgendermaßen aus: Die Kommission schließt die Analyse der Dokumente ab (mit Ausnahme von Medizinprodukten der 1. Risikoklasse und Medizinprodukten für die In-vitro-Diagnostik). Anschließend senden sie ein Schreiben und geben 50 Arbeitstage Zeit, um klinische Studien durchzuführen und die Ergebnisse bereitzustellen. Bei den meisten unserer Gele wurde die gesamte klinische Praxis bereits vor Verschärfung der Anforderungen durchgeführt, sodass keine Schwierigkeiten auftraten. Aber jetzt haben wir eine Zielbestellung von Augenärzten für das neue Blefarogel Forte - es ist wie Blefarogel-2 mit Schwefelverbindungen für Demodex, nur für fortgeschrittene Fälle. Wenn Blefarogel-1 "damit meine Augen nicht weh tun, während ich am Computer sitze", ist Blefarogel-2 "damit es keine Entzündung durch die Zecke gibt und er nichts für immer zu essen hat", dann ist Blefarogel-Forte "Zeckenvölkermord um jeden Preis". In dem Sinne, dass wir das Allergierisiko absichtlich erhöhen, indem wir den Wirkstoffanteil erhöhen, aber die Formel neu kombinieren, damit sie in besonders schwierigen Fällen hilft. Da dies auf Wunsch von Ärzten geschieht, die nur damit rechnen, dass sie ein solches Werkzeug benötigen.
Ende des Epos
Als nächstes wird in Roszdravnadzor die Vollständigkeit der Dokumente überprüft: Ein Unternehmensberater, der einfach das Paket überprüft, fällt auf. Dokumente werden gescannt und an eine Expertenorganisation in der Abteilung von Roszdravnadzor gesendet. Infolgedessen wird in Roszdrav auf der Grundlage der Gesamtheit aller Dokumente eine Schlussfolgerung gezogen. Macht es zu einer Expertengruppe. Wenn alles in Ordnung ist und die Dokumente einander entsprechen, geben sie die Erlaubnis zur staatlichen Registrierung.
Von diesem Moment an können Sie überhaupt nichts mehr anfassen. Ersetzen Sie den Buchstaben auf dem Etikett oder ersetzen Sie das Symbol automatisch. Dies bedeutet, dass Sie sich erneut registrieren müssen. Wir wurden gebeten, das Label von Mediagel-s irgendwie in Russisch-Englisch zu ändern. Dies bedeutet eine erneute Registrierung. Sechs Monate dauerte es, bis der englische Text eingegeben wurde.
Da der Prozess ein Experte ist, ist er weitgehend mit spezifischen Interpretationen ziemlich komplexer Normen verbunden. Dies bedeutet, dass Sie Dokumente mehrmals nach demselben Schema einreichen können. Dann stellt sich heraus, dass ein neues Blatt Papier benötigt wird. Im Allgemeinen ist dies eine Art Unfall: Sie können bei jeder erneuten Einreichung abschließen. Gleichzeitig bleibt die alte staatliche Registrierung bestehen und verschwindet nicht, aber wir verlieren Geld für Gerichtsverfahren und zahlen selbst für die Registrierung und Änderungen eine hohe staatliche Abgabe. Und wir geben Staatsmäuse aus.
Als nächstes müssen Sie auf die Bestellung auf Papier warten. Wenn eine Bestellung eingeht, wird das Produkt in die Registrierung, dh in die
Website, auf der die Apotheken nachsehen, was was ist, abgelegt. Nur von diesem Moment an können Sie etwas tun. Die Apotheke nimmt keine Produkte an, die nicht in der EVU des Gesundheitsministeriums enthalten sind, wenn es sich natürlich nicht um Kosmetika handelt.
Erst in diesem Moment kann die Produktion beginnen. Ohne die Nummer des Gesundheitsministeriums können wir in Russland nicht nur verkaufen, sondern sogar produzieren.
Alles, von diesem Moment an machen wir das Medizinprodukt in Serie. Aus Erfahrung: Es ist sehr wichtig, ein gut ausgebautes Qualitätskontrolllabor zu haben, da jede Beschwerde eine gründliche Überprüfung im Werk bedeutet (gemessen am Verhalten können Sie nicht ohne Bericht über mindestens einige Verstöße zurückkehren) und den Versand einstellen. In diesem Zusammenhang ziehen wir es vor, Abnahmetests und regelmäßige Tests selbst durchzuführen und die Studien an kleinen Proben mit unseren eigenen abzuschließen. Ich meine, 10 Freiwillige aus dem Standard sind wenige, aber 100 Freiwillige sind bereits eine mehr oder weniger repräsentative Stichprobe. Auf lange Sicht zahlt sich eine umfangreichere Forschung immer aus, und für mich ist es ein großes Rätsel, warum andere Hersteller dies nicht immer tun.
Im nächsten Beitrag werde ich versuchen, über eine weitere wichtige Forschungsstufe zu sprechen - den Challenge-Test: In diesem Fall sind die Proben speziell mit pathogenen Bakterien kontaminiert. Dieser Teil der Forschung sollte niemals in der Nähe von Produktions- oder Forschungslabors durchgeführt werden, daher werden die Herausforderungstests durchgeführt.