Proteine spielen in allen lebenden Organismen eine entscheidende Rolle und erfüllen viele verschiedene Funktionen. Wie Sie wissen, bestehen sie aus Aminosäuren. Um ihre Funktionen zu erfüllen, müssen Proteine nicht nur eine Kette bestimmter Aminosäuren sein, sondern eine bestimmte räumliche Form haben, dh richtig in den Raum passen. Aus verschiedenen Gründen kann eine Fehlfunktion bei der normalen Faltung des Proteins in die gewünschte Struktur auftreten. Anstelle von falsch gefalteten Proteinen, die dazu neigen, sich in Clustern zu vereinigen, sind Proteinaggregate Amyloidfibrillen. Das bekannteste dieser Aggregate ist β-Amyloid (Aβ, Abeta), das vermutlich mit der Entwicklung von Neuropathologien sowie einigen Krebsarten und einer der Ursachen für Demenz bei Menschen mit Down-Syndrom assoziiert ist.
Solche Proteinstrukturen haben einen Durchmesser von etwa 5–10 nm und eine Länge von bis zu 800 nm und bestehen aus zwei oder mehr parallelen multidirektionalen Filamenten, die eine spezifische Struktur bilden - die Cross-Beta-Fold-Konformation. Es ist diese Struktur, die die spezifische optische Eigenschaft von Amyloid bestimmt - die Fähigkeit zur Doppelbrechung. Die Entdeckung dieser Eigenschaft ist die Grundlage für die Diagnose einer Amyloidose. Die Mikroskopie von mit Kongorot-Farbstoffpräparaten gefärbten Amyloiden mit polarisiertem Licht ändert die rote Farbe in ein grünes Leuchten [1].
Akkumulatoren abnormaler Proteine werden von den Autoren des SENS-Konzepts „extrazelluläre Trümmer“ (extrazellulärer Müll) genannt und bestimmen eine der Ursachen des Alterns, was ziemlich fair erscheint. Amyloidfibrillen unterliegen aufgrund ihrer Struktur nicht der Wirkung spezieller Enzyme, die Proteine (Proteasen) abbauen, und haben daher die Eigenschaft, sich im Gewebe des Körpers anzusammeln und ihre Arbeit zu stören. Die strukturellen und chemisch-physikalischen Eigenschaften von Amyloid hängen vom Hauptvorläuferprotein ab, dessen Gehalt in der Fibrille etwa 80% beträgt, und dies bestimmt ein spezifisches Merkmal für jede Art von Amyloidose. Der Begriff Amyloidose bezieht sich auf eine Gruppe von erblichen oder erworbenen Krankheiten, die mit der extrazellulären Ablagerung von Fibrillen unlöslicher Proteine verbunden sind, die Gewebestrukturstörungen und Organfunktionsstörungen verursachen. Derzeit sind mehr als 20 amyloidogene Vorläuferproteine und die gleiche Anzahl klinischer Varianten der Amyloidose bekannt. Neben dem bekannten β-Amyloid gibt es AA-Amyloid, das mit rheumatoider Arthritis, Herzerkrankungen, Nierenerkrankungen und Darmentzündungen assoziiert ist, AIAPP-Amyloid, das an der Pathogenese von Typ-2-Diabetes mellitus beteiligt ist, und andere [2].
 β-Amyloid.
β-Amyloid.SENS-Autoren sehen einen Weg, um das Problem der Akkumulation von Proteinaggregaten bei der Verwendung spezialisierter katalytisch aktiver Antikörper, der sogenannten Abzyme (englisches Enzym, Antikörperenzym), die speziell für Amyloide ausgewählt wurden, zu lösen und aus dem Gewebe zu entfernen. Kürzlich wurde eine vielversprechende Methode als Teil dieses Ansatzes entwickelt. Es wurde eine Untergruppe menschlicher Antikörper entdeckt, die eine katalytische Aktivität gegen ein bestimmtes Antigen aufweisen und es in kleinere und weniger schädliche Fragmente zerlegen, anstatt es zur Entfernung oder Zerstörung durch andere Immunzellen einzufangen. Die Verwendung dieser neuen katalytischen Antikörper als Amyloid-Targeting-Therapien bietet potenzielle Vorteile gegenüber der Sequestrierung von Antikörpern, die in anderen Amyloid-Impfstoffen verwendet werden. Das erste ist, dass eine Dosisreduktion erforderlich ist, um extrazelluläre Aggregate effektiv aus Geweben zu entfernen. Dies liegt daran, dass sequestrierende Antikörper jeweils nur ein Amyloidmolekül einfangen und dann transportieren können. Während die Abzyme an das Amyloidmolekül binden, mahlen Sie es und fahren dann mit dem nächsten fort, sodass jedes Antikörpermolekül schnell mehrere Amyloidmoleküle zerstören kann. Ein weiterer Grund ist, dass katalytische Antikörper zu einer Klasse gehören, die effizienter durch die Blut-Hirn-Schranke transportiert wird, die unser Gehirn schützt, während die Sequestrierung von Antikörpern schwieriger ist, diese Schranke zu überwinden [3].
Das bekannteste und am besten untersuchte Amyloid ist heute das β-Amyloid, das viele Forscher der Alzheimer-Krankheit zuschreiben. Diese Pathologie wurde vor mehr als einem Jahrhundert von A. Alzheimer beschrieben, der als erster auf das pathomorphologische Hauptsymptom dieser Krankheit hinwies - unlösliche senile Plaques im Gehirn der Toten, die an dieser Krankheit litten. Heute gibt es weltweit mehr als 40 Millionen Menschen mit einer Alzheimer-Diagnose - die Wahrscheinlichkeit, diese Pathologie zu entwickeln, verdoppelt sich nach 65 Jahren alle fünf Jahre. Und die langfristigen Schätzungen der WHO bezüglich des massiven Anstiegs solcher Patienten in den kommenden Jahrzehnten sind äußerst pessimistisch.
Es ist bekannt, dass unlösliche Plaques im Gehirn von Patienten hauptsächlich durch β-Amyloidpeptid (Aβ) gebildet werden, das ein Molekulargewicht von 4 kDa und eine Länge von etwa 40 Aminosäureresten aufweist. Aβ ist ein Fragment des Transmembranproteins des Amyloid-Vorläuferproteins (Amyloid-Vorläuferprotein, APP), das in vielen Geweben des Körpers, einschließlich in den Synapsen von Neuronen, gefunden wird. APP ist an einigen physiologischen Prozessen beteiligt, die mit Neuroplastizität, Synapsenbildung und Neuroprotektion (Überleben von Nervenzellen) verbunden sind [4].
Im Gegensatz zu seinem Vorgänger ist Aβ für Nervenzellen toxisch und trägt zu deren Degeneration und Tod bei. Es wird durch Trennen der extrazellulären N-terminalen Domäne (sAPP) des Vorläuferproteins gebildet. Dieser Prozess kann von zwei verschiedenen Sekretasen durchgeführt werden - α-Sekretase und β-Sekretase, die einen grundlegenden Unterschied in ihrer Wirkung aufweisen. Im ersten Fall tritt eine Fragmentierung zwischen Aminosäureresten innerhalb der Aβ-Sequenz auf, was die nachfolgende Bildung eines Amyloidpeptids verhindert. Zweitens endet pathologisch unter dem Einfluss der β-Sekretase der Fragmentierungsprozess aufgrund seiner Merkmale in der Bildung von Aβ. Dieser zweite Weg, der mit der Entwicklung von Neuropathologien verbunden ist, ist seltener und warum die Fragmentierung des Vorläuferproteins folgt, bleibt nicht vollständig klar [5].
 β-Sekretase.
β-Sekretase.Die Neurotoxizität von Aβ ist mit einer Verletzung der Calciumhomöostase, der Exzitotoxizität, entzündlichen Prozessen, der Stimulation von oxidativem Stress und der Apoptose verbunden. Die vorherrschende Meinung war, dass die Akkumulation von Aβ im Gehirn ein rein pathologischer Prozess ist, da es unmöglich ist, Aβ aus dem Gehirngewebe zu entfernen. Heute ist bekannt, dass Aβ dennoch auf mindestens zwei Arten aus dem Gehirn eliminiert werden kann: perivaskulär (durch Lymphe) und proteolytisch (Spaltung durch verschiedene Enzyme) [6, 7]. Daher kann die Bildung der Alzheimer-Krankheit im Alter mit der damit einhergehenden Anreicherung unlöslicher Amyloidfibrillen mit verschiedenen stimulierenden Faktoren verbunden sein. Dies kann genetischer Natur sein und mit Stress, Hypoxie, Ischämie und früheren Schlaganfällen verbunden sein [8].
β-Amyloid oxidiert Cholesterin und mehrfach ungesättigte Fettsäuren und bildet die giftigsten Formen reaktiver Sauerstoffspezies - Hydroxylradikale und Wasserstoffperoxid. Die Racemisierung von L-Asparaginsäure in langlebigen Proteinen fördert die Bildung von β-Amyloid und α-Synuclein. Pathologische Ansammlungen der letzteren werden bei Parkinson, Alzheimer, Levy und anderen neurodegenerativen Erkrankungen beobachtet.
Ein weiteres pathomorphologisches Zeichen der Neurodegeneration bei Alzheimer sind Aggregate von hyperphosphoryliertem Tau-Protein (τ-Protein): gepaarte helikale Filamente (PHF) und neurofibrilläre Verwicklungen (NFT). Physiologisch ist das τ-Protein an der Stabilisierung von Mikrotubuli von Neuronen beteiligt, die den Transfer von zellulären Organellen, Glykoproteinen und anderen Substanzen durch das Zytoplasma von Neuronen sicherstellen. Bei der Alzheimer-Krankheit ist das τ-Protein hyperphosphoryliert, verliert seine normale Fähigkeit zur Stabilisierung von Mikrotubuli und reichert sich in der Zelle mit unlöslichen neurotoxischen Strukturen an. Was früher während der Bildung der Pathologie passiert - die Bildung von Amyloid- oder τ-Proteinaggregaten - ist keine klare Frage. Aber offensichtlich sind diese beiden Prozesse miteinander verbunden und stimulieren sich gegenseitig. Beide pathologischen Proteine weisen also ähnliche Prion-Eigenschaften wie das PrPsc-Prion-Protein auf: Falsch gefaltete Formen von Proteinen durch die Art der Kettenreaktion stimulieren die Umwandlung normaler Proteine in unregelmäßige in gesunden Neuronen, die sie umgeben. Aβ und das τ-Protein interagieren untereinander auch durch die Art der Prionen: Es wird beschrieben, wie Aβ die GSK3-Proteinkinase aktiviert, indem es das τ-Protein phosphoryliert und dessen unsachgemäße Bildung bewirkt [5].
 τ Protein.
τ Protein.In jüngerer Zeit wurde angenommen, dass Prionkrankheiten und Alzheimer-Krankheit zwar bestimmte biochemische Ähnlichkeiten aufweisen, die zweite Pathologie jedoch im Gegensatz zur ersten nicht infektiös ist und nicht von einem Organismus auf einen anderen übertragen wird. Aber im Jahr 2015 erschienen die ersten beunruhigenden Nachrichten. In der Zeitschrift Nature wurde von britischen Neurologen ein Artikel veröffentlicht, in dem sie über den möglichen Weg der Aβ-Übertragung von einer Person zur anderen sprachen. Die Autoren führten eine Autopsiestudie des Gehirns von acht Personen durch, die an der Creutzfeldt-Jakob-Krankheit starben. Bei den sechs Toten wurden zusätzlich zu den mit der Grunderkrankung verbundenen Verletzungen umfangreiche Ansammlungen von Amyloiden im Gehirn gefunden. Was weder für das junge Alter der Toten noch für diese Pathologie untypisch ist. Es wurden auch keine Defekte im Zusammenhang mit Amyloidogenese und Neuropathologien in ihren Genomen gefunden. Die Forscher schlugen vor, dass Aβ während der Injektion von mit Amyloid infiziertem Wachstumshormon in den Körper des Verstorbenen eingeführt wurde. Genauso wie mehrere hundert Menschen zuvor infiziert waren und an Injektionen von wachstumshormonhaltigen Prionen starben [9].
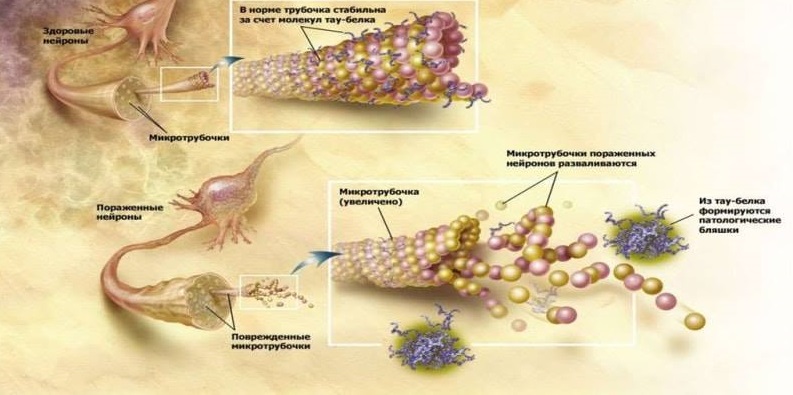 Die Funktionen des τ-Proteins sind normal und pathologisch.
Die Funktionen des τ-Proteins sind normal und pathologisch.Die Bestätigung der Infektiosität der Alzheimer-Krankheit kann eine weitere Studie sein, in der Labormäuse mit erhöhter Amyloidogenese und normale Mäuse chirurgisch mit dem Blutfluss kombiniert wurden. Infolgedessen begannen gesunde Mäuse, Aβ im Gehirn anzusammeln, was für diese Nagetiere nicht typisch ist. Diese Studie zeigte erstmals das Potenzial für die Penetration von Aβ mit Blut in das Gehirn und die anschließende Beteiligung an der Entwicklung der Neurodegeneration [10].
Die Beziehung zwischen der Akkumulation von β-Amyloid und τ-Protein in Geweben mit altersbedingten Neuropathologien bestimmt die Notwendigkeit, nach wirksamen Methoden zu suchen, um diese Proteinaggregate als Biomarker für die zukünftige Neuropathologie und das beschleunigte Altern zu identifizieren. Einige solcher Methoden sind bereits heute bekannt.
Amyloidablagerungen im Gehirngewebe können mittels Positronenemissionstomographie nachgewiesen werden, wenn β-Amyloid durch das in den Körper eingebrachte und mit dem Amyloid verbundene radioaktive Isotop bestimmt wird.
Einer der empfindlichsten Biomarker bei der Früherkennung von Alzheimer und mittelschweren Kongestivstörungen (UKI) beim Übergang in die Pathologiephase wird heute als der Gehalt an Amyloid Aβ-42, Gesamt-τ-Protein und phosphoryliertem τ-Protein in der Cerebrospinalflüssigkeit (CSF) angesehen. Darüber hinaus zeigen Amyloide mit Pathologie eine Abnahme des Spiegels, und die Spiegel in der Cerebrospinalflüssigkeit des τ-Proteins, insgesamt und phosphoryliert, nehmen zu. Dies ist auf die Tatsache zurückzuführen, dass bei gesunden Menschen keine Ansammlungen von Amyloid in Form von Plaques vorliegen und daher eine große Menge an freiem Amyloid in der Cerebrospinalflüssigkeit gefunden wird. Niedrige Werte des τ-Proteins zeigen das Fehlen einer Zerstörung des neuronalen Zytoskeletts.
Es wurde eine signifikante Korrelation zwischen dem Alter und einer Verlangsamung der Ausscheidung von β-Amyloid aus dem Nervensystem gefunden, was mit Sklerose der Lymphgefäße und einer Abnahme ihrer Drainagefähigkeit verbunden sein kann. Die Wiederherstellung der Drainage ist eine der vielversprechenden Behandlungen für die Alzheimer-Krankheit, die von Leucadia vorgeschlagen wurde. Ein Interview mit dem Gründer wurde übersetzt und auf
habr.com/post/371513 veröffentlichtDaher kann die Konzentration Aβ-42 in der Cerebrospinalflüssigkeit in Höhe von 716,9 ± 94,2 ng / ml als normaler Indikator für eine gesunde Person angesehen werden. Und der Aβ-42-Spiegel, der ein erhöhtes Risiko für Neuropathologie zeigt, liegt bei <209 ng / ml. Für ein τ-Protein gelten Werte von 73,9 ± 51,7 ng / ml als Normalwerte in der Cerebrospinalflüssigkeit. Ein Anstieg der τ-Proteinspiegel auf 231,6 ± 158,5 ng / ml kann bereits auf das Vorhandensein eines aktiven neurodegenerativen Prozesses hinweisen. Nach den heutigen Vorstellungen zeigt eine kombinierte Untersuchung der Aβ-42- und τ-Proteinspiegel in der Liquor cerebrospinalis eine hohe Empfindlichkeit bei der Früherkennung von Neuropathologien - 94% der Wahrscheinlichkeit, Alzheimer zu diagnostizieren [11].
Bald wird das Aufkommen ultraempfindlicher immunologischer und massenspektrometrischer Methoden zur Diagnose von Amyloidose (Verhältnisse von Aβ42 / 40- oder APP669-711 / Aβ42-Proteinen) und Neurodegeneration (τ-Proteine und Neurofilamente) im Blutplasma erwartet [12]. Es gibt auch „marklose“ Methoden zum Nachweis von falsch gefalteten Proteinen im Plasma [13].
Autoren der Rezension: Denis Odinokov, Alexey Rzheshevsky.
Liste der verwendeten Literatur- Rameev V., Kozlovskaya L. Amyloidose: moderne Diagnose- und Behandlungsmethoden. Effektive Pharmakotherapie. Urologie und Nephrologie. 2012. Nr. 11, S. 6-15.
- Butler L. I., Karpova O.Yu., Alexandrova E.N., Petrova S.Yu. Amyloidose des Herzens bei älteren Menschen. Archiv für Innere Medizin. 2015. Nr. 6 (26), p. 28-36.
- AmyloSENS: Entfernen von Müll zwischen Zellen.
- 4Lee, V., Goedert, M., Trojanowski, J. (2001) Neurodegenerative Tauopathies, Annu. Rev. Neurosci., 24, 1121–1159.
- Tatarnikova O.G., Orlov M.A., Babkova N.V. Amyloid Beta und Tau Protein: Struktur, Wechselwirkung und prionähnliche Eigenschaften. Fortschritte in der biologischen Chemie, Bd. 55, 2015, S. 351-390.
- Weller, R., Yow, H., Preston, S., Mazanti, I., Nicoll, J. (2002) Zerebrovaskuläre Erkrankungen sind ein Hauptfaktor für das Versagen der Elimination von Amyloid Beta aus dem alternden menschlichen Gehirn, Ann. NY Acad. Sci., 977, 162–168.
- N. Nalivaeva, L. Fisk, Belyaev. N., Turner, A. (2008) Amyloid abbauende Enzyme als therapeutische Ziele bei Alzheimer, Curr. Alzheimer Res., 5, 212–224.
- L. Fisk, N. Nalivaeva, J. Boyle, C. Peers, A. Turner (2007) Auswirkungen von Hypoxie und oxidativem Stress auf die Expression von Neoprilysin in menschlichen Neuroblastomzellen und kortikalen Neuronen und Astrozyten von Ratten, Neurochem. Res. 32, 1741 & ndash; 1748.
- Jaunmuktane Z., Mead S., Ellis M., Wadsworth JDF, Nicoll AJ, Kenny J. et al. (2015). Hinweise auf eine Übertragung der Amyloid-β-Pathologie und der cerebralen Amyloid-Angiopathie beim Menschen. Nature 525, 247 & ndash; 250.
- XL Bu, Y Xiang, WS Jin, J Wang, LL Shen, ZL Huang, K Zhang, YH Liu, F Zeng, JH Liu, HL Sun, ZQ Zhuang, SH Chen, XQ Yao, B Giunta, YC Shan, J Tan , XW Chen, ZF Dong, HD Zhou, XF Zhou, W Song und YJ Wang. Aus Blut stammendes Amyloid-β-Protein induziert Pathologien der Alzheimer-Krankheit. Molekulare Psychiatrie. 2017.
- V.Yu. Lobzin, A.Yu. Emelin, L.A. Alekseeva. Liquorologische Biomarker der Neurodegeneration bei der Früherkennung kognitiver Beeinträchtigungen. Bulletin der Russischen Militärakademie. 2013, No. 4, p. 15-20.
- Blennow, Kaj und Henrik Zetterberg. "Biomarker für die Alzheimer-Krankheit - aktueller Stand und Perspektiven für die Zukunft." Zeitschrift für Innere Medizin (2018).
- Nabers, Andreas et al. "Amyloid-Blut-Biomarker erkennt Alzheimer." EMBO Molecular Medicine (2018).