Mitochondrien - kleine Arbeiter oder große Chefs?Wenn Sie der Meinung sind, dass die wichtigste Geschichte für das Zusammenleben während der Hochzeit beginnt, ist dies überhaupt nicht der Fall. Die wichtigste Geschichte im Leben eines jeden Menschen begann vor mehr als einer Milliarde Jahren, als unsere entfernten einzelligen Vorfahren gezwungen waren, einen „Ehevertrag“ mit denen zu schließen, die wir heute Mitochondrien nennen (siehe Theorie der Symbiogenese).
Mitochondrien haben zwei Membranen (intern und extern) und ein eigenes Erbmaterial in Form von DNA (Abb. 1). Auf der inneren Membran der Mitochondrien befindet sich ein oxidatives Phosphorylierungssystem, dessen Betrieb die Oxidation von Energiesubstraten unter Bildung von ATP ermöglicht.
Abb. 1 . Schematische Struktur der Mitochondrien
Im Ehevertrag von Zelle und Mitochondrien gibt es keine Klausel "in Krankheit und Gesundheit" - und gut. Wenn die Mitochondrien alt werden, kann die Zelle sie während der Mitophagie abtöten, und die Mitochondrien regulieren wiederum den Apoptoseprozess in dysfunktionellen und alten Zellen. Wenn der Prozess der gegenseitigen Qualitätskontrolle gestört wird, werden Alterungsmechanismen eingeleitet. Die Mechanismen der Apoptose werden gestört, die Anzahl der freien Radikale, die nicht von Mitochondrien kontrolliert werden, nimmt zu. Dies führt zu systemischen Entzündungen und Schäden an der DNA der Zelle. Somit besteht ein enger Zusammenhang zwischen MX-Dysfunktion, altersbedingten Erkrankungen, Alterung und Stoffwechselstörungen [1]. Stoffwechselstörungen sind ein ständiger Treiber der Apokalypse des Alterns.
"Wie ein Eichhörnchen in einem Rad" - die Dynamik der Mitochondrien
Nicht alle Schuld an Stoffwechselstörungen liegt in unserem übermäßigen Essen. Stoffwechselstörungen sind in erster Linie mit der Unfähigkeit der Mitochondrien verbunden, mit Nährstoffen umzugehen. Mitochondrien in der Zelle sind nicht einfach. Wir „füttern“ unsere Zellen entweder zu viel oder zu wenig und stellen ihnen eine „Aufforderung“, Energie in Form von ATP abzugeben, deren Menge genau unseren Bedürfnissen entsprechen muss. Um regelmäßig aus dieser Situation herauszukommen, verwenden Mitochondrien wirklich einige „Bewegungen“ - Spaltung und Fusion. Diese „Mitodomotion“ werden unter dem Namen „Mitochondriendynamik“ zusammengefasst. Das Gleichgewicht zwischen mitochondrialer Teilung und Fusion ist der zentrale Mechanismus der bioenergetischen Anpassung an die Stoffwechselbedürfnisse der Zelle [2, 3].
Die meisten Mitochondrien befinden sich in Geweben mit hohem Energiebedarf - Muskeln, Leber, braunes Fettgewebe und Gehirn. Es ist nicht überraschend, dass die Dynamik der Mitochondrien in diesen Geweben besser untersucht wurde.
Wenn also eine Zelle eines dieser Gewebe (mit Ausnahme einiger Neuronen im Gehirn, dazu später mehr) eine große Menge an Nährstoffen erhält (die Aufnahme übersteigt die Kosten), befinden sich die Mitochondrien in einem geteilten (fragmentierten) Zustand. Befindet sich die Zelle in einem Hungerzustand (Einkommen geringer als die Kosten), verschmelzen die Mitochondrien und sie befinden sich in einem verbundenen Zustand. [3,4]. Auf diese Weise wird die Zellhomöostase aufrechterhalten (Abb. 2).
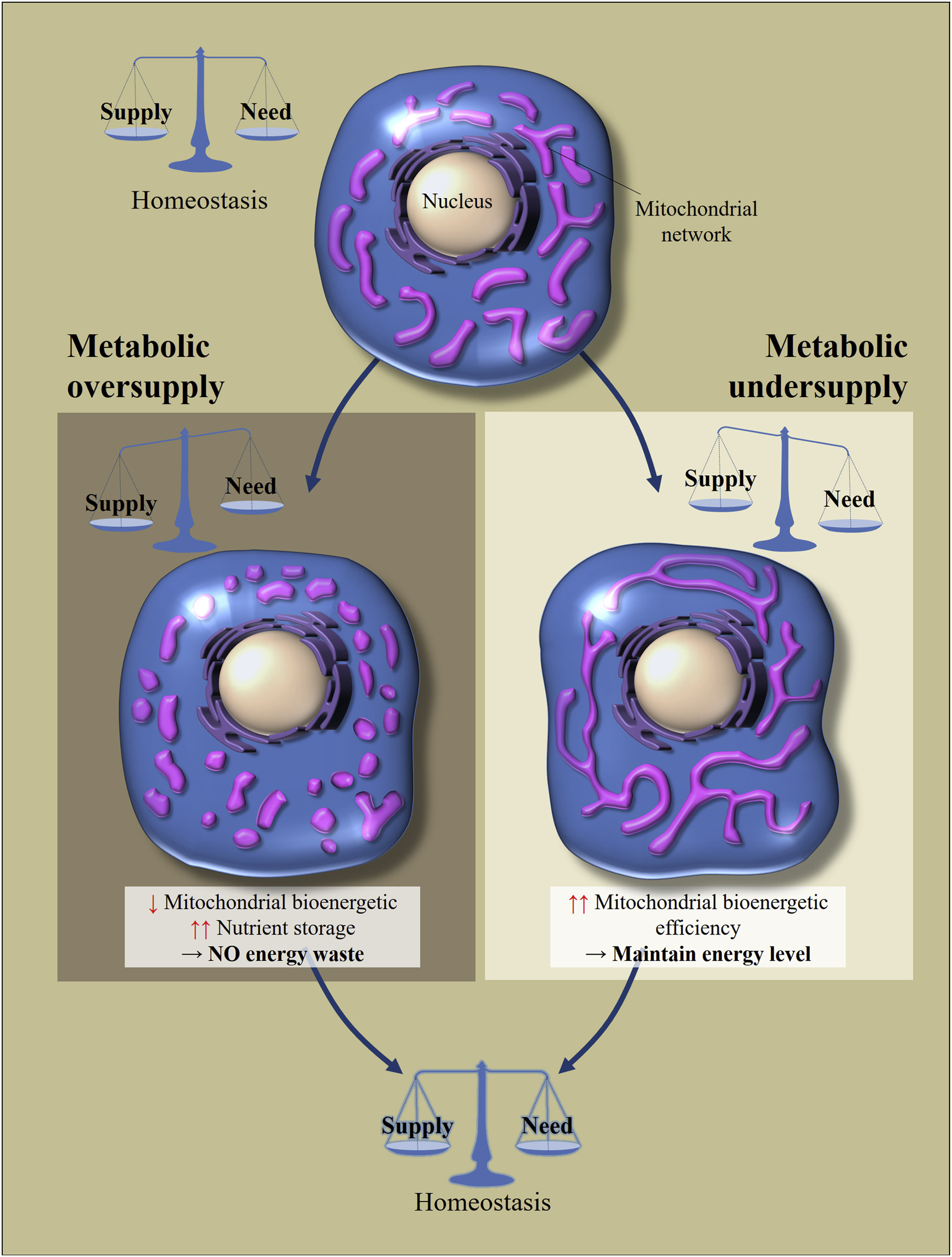 Abb. 2
Abb. 2 Regulation der Morphologie und Bioenergieeffizienz von Mitochondrien als Reaktion auf übermäßige oder unzureichende Nährstoffaufnahme [von 2]
Die zelluläre metabolische Homöostase hängt vom Gleichgewicht zwischen Nährstoffaufnahme und -verbrauch ab. Änderungen in der Nährstoffversorgung führen zu zellulären Anpassungen, um das Gleichgewicht wiederherzustellen. Übermäßige Ernährung führt zu einer Fragmentierung des mitochondrialen Netzwerks, was zu einer Verringerung der bioenergetischen Effizienz der Mitochondrien führt. Dies vermeidet Energieverluste. Im Gegensatz dazu werden Mitochondrien mit metabolischem Hunger länger, um ihre bioenergetische Effizienz zu erhöhen.Was ist der Trick dieser Bewegungen? Wenn die Zelle in einem Hungerzustand ist, kann die Fusion von Mitochondrien ihre Bioenergieeffizienz erhöhen (die Menge an ATP, die pro Nährstoffmolekül erzeugt wird). Wenn ein Überschuss an Nährstoffen in die Zelle gelangt, können diese entweder 1) gespeichert oder 2) diese Energie in Form von Wärme abgeführt werden. Die Aufgabe der Mitochondrien besteht in diesem Fall darin, mehr Energie in Form von Wärme abzuleiten und weniger in Form von ATP zu speichern (die Akkumulation von NADH und ROS führt zu oxidativem Stress). Die Fragmentierung von Mitochondrien ermöglicht es ihnen, die Bioenergieeffizienz zu verringern, deren Hauptmechanismus der Reduktion als "Leckage" von Protonen angesehen wird.
Also machen wir uns an die Arbeit und das Leben der Mitochondrien verläuft ständig im Kreislauf von Teilung und Fusion (Abb. 3).
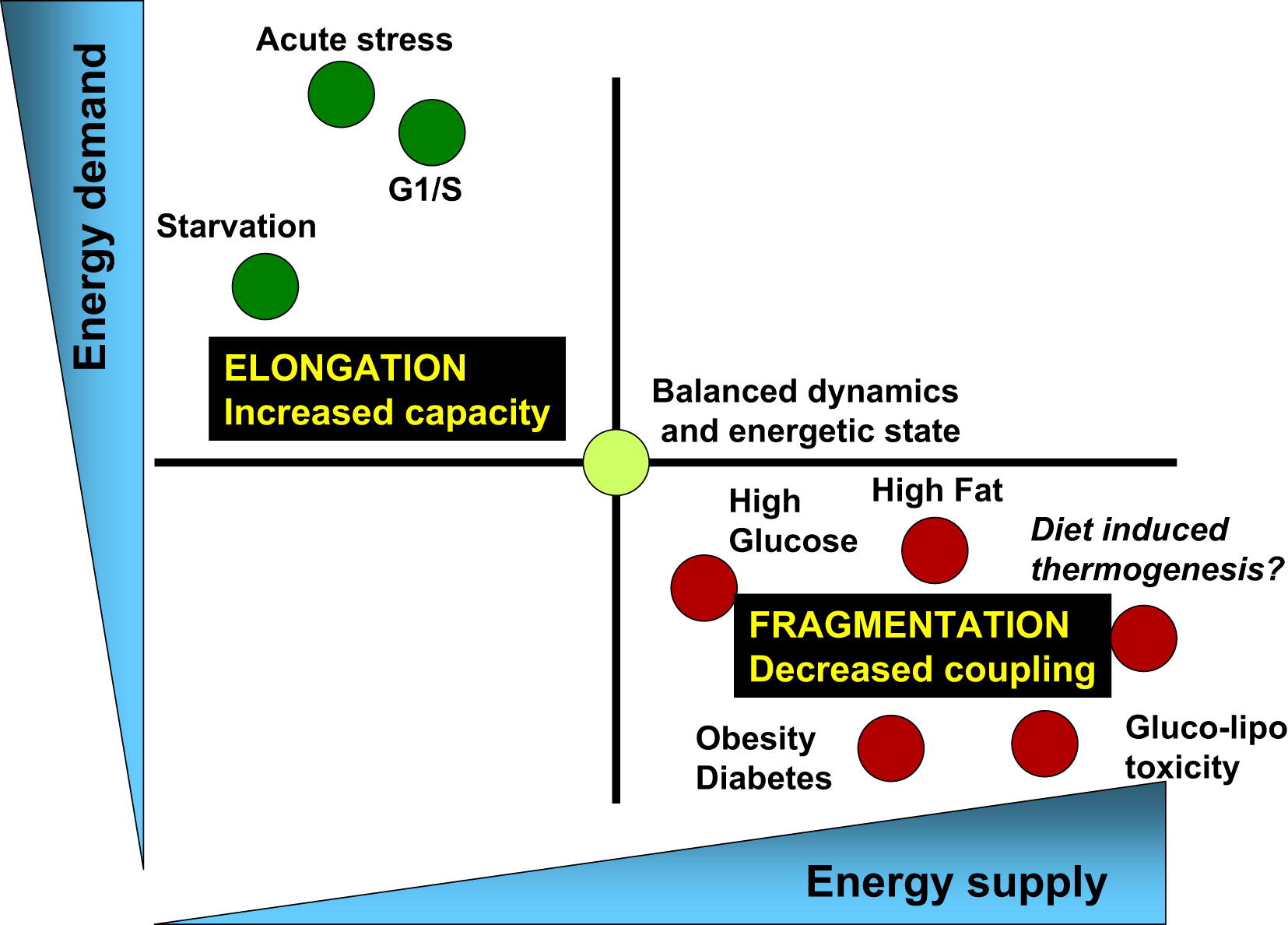 Abb. 3 Das Gleichgewicht zwischen Energieverbrauch und Energieversorgung ist mit entsprechenden Änderungen in der Architektur der Mitochondrien und ihrer Bioenergieeffizienz verbunden [von 3].
Abb. 3 Das Gleichgewicht zwischen Energieverbrauch und Energieversorgung ist mit entsprechenden Änderungen in der Architektur der Mitochondrien und ihrer Bioenergieeffizienz verbunden [von 3].
Die physiologischen Prozesse, die mit einem Anstieg des Energiebedarfs und einer Abnahme der Energieversorgung verbunden sind (z. B. akuter Stress, Hunger und die G1 / S-Phase), sind durch eine Verlängerung der Mitochondrien und Atmung im Zusammenhang mit der ATP-Synthese gekennzeichnet. Andererseits sind physiologische Prozesse, die mit einer Abnahme des Energiebedarfs und einer Zunahme seines Angebots verbunden sind (hohe Nährstoffkonzentrationen, Fettleibigkeit und Typ-2-Diabetes), mit einer Fragmentierung der Mitochondrien, einer Wärmeerzeugung oder einer verminderten Mitochondrienfunktion verbunden.Gesunde Spalt- und Fusionszyklen sind der Schlüssel zur Gesundheit des Zellstoffwechsels
Der normale Zyklus der mitochondrialen Teilung und Fusion ist ein Schlüsselelement bei ihrer Qualitätskontrolle. Warum? Während der mitochondrialen Teilung werden zwei Tochtergesellschaften gebildet, von denen eine ein höheres Membranpotential aufweist und weiter in den Fusionsteilungszyklus übergeht, und die andere mit einer stärker depolarisierten Membran bleibt getrennt, bis das Membranpotential wiederhergestellt ist. Wenn das Potential wiederhergestellt ist, vereinigt es sich wieder mit dem mitochondrialen Netzwerk. Bleibt es depolarisiert, wird es bei der Autophagie eliminiert, was der Schlüssel zur Qualität des Mitochondrienpools ist (Abb. 4).
Eine langfristige Hemmung der Mitochondrienteilung (mit längerem Zellmangel) führt zur Akkumulation geschädigter Mitochondrien, die nicht getrennt werden können [3, 4].
Andererseits führt ein Überschuss an Nährstoffen zu einer Hemmung der Mitochondrienfusion, was zu einer Störung des mitochondrialen Dynamikzyklus führt und die intrazelluläre mitochondriale Heterogenität erhöht. Ja, bei einem Überschuss an Nahrungsmitteln ist die Fragmentierung der Mitochondrien schützend, aber eine längere Fragmentierung ist ebenso wie eine längere Fusion schädlich für die Qualitätskontrolle der Mitochondrien. Es erfolgt keine selektive Entfernung, die mitochondriale Masse nimmt ab und besteht aus kleinen depolarisierten Mitochondrien.
 Abb. 4 Lebenszyklus von Mitochondrien und ihre Regulierung der Nährstoffverfügbarkeit [von 3]
Abb. 4 Lebenszyklus von Mitochondrien und ihre Regulierung der Nährstoffverfügbarkeit [von 3]Mitofusine sind nicht irgendwelche Proteine
Auf molekularer Ebene ist die Mitochondrienfusion ein zweistufiger Prozess, der eine koordinierte Fusion der äußeren und inneren Membran während getrennter aufeinanderfolgender Ereignisse erfordert. Bei Säugetieren wird dieser Prozess durch drei Proteine reguliert, die zu GTPasen gehören: Mfn1 und Mfn2 sind für die Fusion der Außenmembran erforderlich, und OPA1 - für die Fusion der Innenmembran. Für die Teilung werden andere Proteine benötigt, Fis1 und Drp1.
Die Rolle von Mitofusin-Proteinen wurde in Funktionsverlust- und Gewinngewinnstudien untersucht. Mäuse, die für Mitofusin-Proteine mutiert sind, sterben bereits in der Mitte der Schwangerschaft, weil eine mitochondriale Fusion für sie unmöglich wird. Mitofusine sind wichtig für die Prozesse der Autophagie und Mitophagie. Eine verminderte Expression von Mfn2 in Kardiomyozyten blockiert den Beginn des Autophagieprozesses, da die Fusion von Autophagosomen mit Lysosomen blockiert ist. Die Abreicherung von Mfn2 führt zu einer Abnahme des Potentials der Mitochondrienmembranen, um dies zu kompensieren, nimmt die Atmungskette ab, die Glukoseaufnahme nimmt zu und die Glykogensynthese nimmt ab. Die Zelle wechselt zur anaeroben Glyozytie, und dies ist der Weg zur onkologischen Degeneration der Zelle. Ein Mfn2-Mangel führt zu neurodegenerativen Veränderungen. Eine Erhöhung der Expression von Mfn2 in den Skelettmuskeln erhöht deren Empfindlichkeit gegenüber Insulin.
Mfn1 erfüllt ähnliche Funktionen, aber wahrscheinlich in anderen Geweben (die Expression von Mfn2 und Mfn1 variiert in verschiedenen Geweben) - Mfn1 wird mehr im Herzen, in der Leber, in der Bauchspeicheldrüse, in den Hoden und in Mfn2 im Herzen, im Skelettmuskel, im Gehirn und im braunen Fettgewebe exprimiert .
Mitofusine sind daher Schlüsselregulatoren der mitochondrialen Dynamik. Die Expression von Mitofusinen ist in verschiedenen Organen unterschiedlich, sie bieten Bioenergieeffizienz und Anpassungsmechanismen an die Verfügbarkeit von Nährstoffen, und das "Schicksal" der Zelle hängt von ihnen ab. Es überrascht nicht, dass mitochondriale Fusionsproteine potenzielle Ziele für pharmakologische Interventionen sind [2, 5].
Hypothalamus, Mitochondrien, Stoffwechselstörungen und Alterung
Die Dynamik der Mitochondrien ist in allen Zellen wichtig. In Beta-Zellen der Bauchspeicheldrüse sind Mitochondrien Nährstoffsensoren und Generatoren von Insulinsynthesesignalen. In Muskeln ist die Dynamik der Mitochondrien wichtig für die Regulierung des Glukosestoffwechsels usw. Eine Person ist jedoch nicht nur eine Sammlung verschiedener Zelltypen, von denen jeder unabhängige Entscheidungen trifft. Ein Organismus ist ein System, das eine zentrale regulatorische Verbindung zur Aufrechterhaltung der Energie- und Glukosehomöostase aufweist. Dieser Hauptregulator ist der Hypothalamus.
Der Hypothalamus befindet sich im Zwischenhirn und ist es, der die Verbindung des nervösen und des humoralen Regulationssystems herstellt. Hypothalamusneuronen nehmen Signale von Fettgewebe (Leptin), Bauchspeicheldrüse (Insulin) und anderen hormonellen Reizen (Ghrelin, Cholecystokinin, Pankreas-Polypeptid usw.) wahr, verarbeiten sie und reagieren darauf. Der Hypothalamus steuert die Aktivität des menschlichen endokrinen Systems aufgrund der Tatsache, dass seine Neuronen neuroendokrine Transmitter sekretieren können, die die Produktion von Hormonen durch die Hypophyse stimulieren oder hemmen. Mit anderen Worten, der Hypothalamus, dessen Masse 5% des Gehirns nicht überschreitet, ist das Zentrum der Regulierung der endokrinen Funktionen und der Aufrechterhaltung der Homöostase des gesamten Organismus.
Sogar Dilman (Dilman V. M "Große biologische Uhr") wies auf die führende Rolle des Hypothalamus bei der systematischen Entwicklung von Stoffwechselstörungen hin, die zu Fettleibigkeit, Diabetes, Herz-Kreislauf-Erkrankungen, onkologischen Erkrankungen und Alterung führen. Nach der von Dilman gebildeten Theorie der Hyperadaptose nimmt die Empfindlichkeit der hypothalamischen Rezeptoren gegenüber Signalen aus Körpergeweben (Leptin, Insulin usw.) mit zunehmendem Alter systematisch ab. Um seine „Reaktion“ zu provozieren, wird immer mehr des einen oder anderen Hormons benötigt - mehr Insulin, mehr Leptin. Entwickelt Insulin- und Leptinresistenz, Stoffwechselerkrankungen, die zu Alterung und Tod führen.
Abhängig von den ausgeführten Funktionen werden Gruppen von Neuronen zu den Kernen des Hypothalamus zusammengefasst. Einer von ihnen - der bogenförmige (bogenförmige) Kern ist ein Schlüsselregulator des Essverhaltens und des Stoffwechsels. Orexigene Neuropeptide (stimulieren den Appetit) und magersüchtige (unterdrücken den Appetit), die AgRP- bzw. POMC-Neuronen entsprechen, können sich darin bilden. Periphere Signale (Insulin, Ghrelin, Leptin usw.) beeinflussen die Expression von Peptiden, die den Appetit anregen oder unterdrücken, wodurch die Kohärenz der zentralen Regulation sichergestellt wird (Abb. 5).
 Abb. 5. Hypothalamische Kontrolle des Energiestoffwechsels. Das Gehirn integriert Stoffwechselsignale (Leptin, Insulin, Ghrelin, PYY3-36) aus peripheren Geweben wie Bauchspeicheldrüse, Fettgewebe und Magen. Im Gehirn koordinieren spezialisierte neuronale Netze adaptive Veränderungen bei der Aufnahme und dem Verzehr von Nahrungsmitteln [von 5].
Abb. 5. Hypothalamische Kontrolle des Energiestoffwechsels. Das Gehirn integriert Stoffwechselsignale (Leptin, Insulin, Ghrelin, PYY3-36) aus peripheren Geweben wie Bauchspeicheldrüse, Fettgewebe und Magen. Im Gehirn koordinieren spezialisierte neuronale Netze adaptive Veränderungen bei der Aufnahme und dem Verzehr von Nahrungsmitteln [von 5].Wer und wie reguliert die Empfindlichkeit von hypothalamischen Neuronen?
Eine Untersuchung der Dynamik von Mitochondrien in Hirngeweben zeigte, dass die Dynamik von Mitochondrien eine wichtige Rolle für die Fähigkeit hypothalamischer Neuronen spielt, den Glukosespiegel und die Energiehomöostase im Körper zu steuern [6,7,8].
In AgRP-Neuronen (hungerfördernde AgRP-Neuronen), die den Appetit anregen und die Gewichtszunahme regulieren, führt Hunger zur Teilung der Mitochondrien und fettreiche Ernährung zur Fusion. Das heißt, die Reaktion der Mitochondrien unterscheidet sich von der in den meisten anderen Zellen.
Die Fusion von MX in diesen Neuronen reguliert die elektrische Aktivität als Reaktion auf eine fettreiche Ernährung und stimuliert die Produktion eines orexigenen Peptids (AgRP-Peptid), das für die Gewichtszunahme und Fettablagerung mit einem Überschuss an Nährstoffen erforderlich ist. Deletionen von Mfn1 und Mfn2 in diesen Neuronen führten bei Ratten aufgrund geringerer Spiegel an zirkulierendem Leptin zu einer geringeren Gewichtszunahme.
POMC-Neuronen (unterdrücken den Appetit) haben die entgegengesetzte Funktion, und die Dynamik der Mitochondrien als Reaktion auf die Nährstoffaufnahme ist unterschiedlich. Eine Abnahme der Expression von Mitofusinen in diesen Neuronen führt zu einer Störung der Verbindung von Mitochondrien mit EPS und infolgedessen zu Hyperphagie, Leptinresistenz und Fettleibigkeit. Gleichzeitig nahm der Lebensmittelverbrauch zu und der Energieverbrauch ab.
Daher hängt die Reaktion des Körpers auf eine fettreiche Ernährung von Mustern der mitochondrialen Dynamik in hypothalamischen Neuronen ab. Der Umbau von Mitochondrien in Neuronen reagiert auf die Nährstoffaufnahme und stimuliert die Produktion von Neuropeptiden, die den Appetit entweder stimulieren oder unterdrücken und den Stoffwechsel auf Körperebene beeinflussen (Abb. 6).
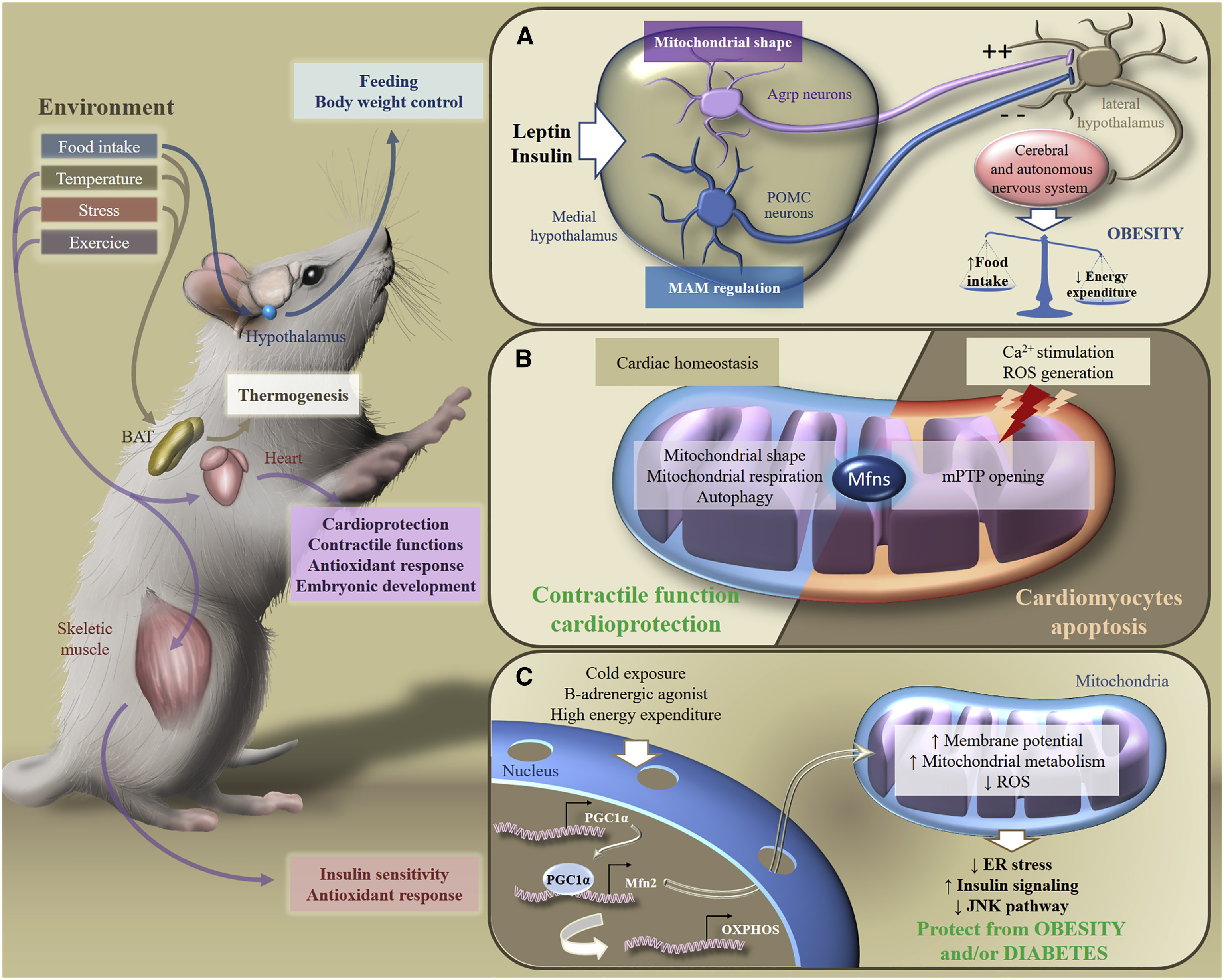 Abb. 6. Stoffwechselanpassung an Umweltreize [von 2]
Abb. 6. Stoffwechselanpassung an Umweltreize [von 2]In Reaktion auf exogene Reize sind Mfns an der Übertragung von Stoffwechselsignalen in verschiedenen Organen beteiligt, wodurch die Aufrechterhaltung der Energiehomöostase im gesamten Körper sichergestellt wird. Insbesondere als Reaktion auf Nahrungsaufnahme, Temperaturänderungen, Stress oder Bewegung passen braunes Fettgewebe, Gehirn, Herz oder Skelettmuskel ihren Stoffwechsel an, um Ernährung, Körpergewicht, kontraktile Funktion, antioxidative Reaktion oder Insulinsensitivität zu steuern.
Wie kann man die Dynamik von Mitochondrien beeinflussen?
1. Ernährung und BewegungErnährungszyklen Überschüssige Nahrung und eine fettreiche Ernährung (HFD) hemmen die Mitochondrienfusion in Zellen (der Mechanismus ist bei einigen Gehirnneuronen unterschiedlich). Ein unvollständiger mitochondrialer Teilungs-Fusions-Zyklus stört Autophagieprozesse → Die intrazelluläre mitochondriale Heterogenität nimmt zu → Es findet keine selektive Entfernung von Mitochondrien statt → Mitochondrien mit akkumulierter Dysfunktion.
Kalorienreduktion (Fütterungs- / Fastenzyklus) stimuliert die bioenergetische Anpassung und bietet mitochondriale Qualitätsmechanismen.
2. Gesunde Membranen: Stearinsäure, Cardiolipin, PhosphatidsäureAlle Schlüsselprozesse hängen von der „Gesundheit“ der Mitochondrienmembranen ab - Autophagie, Mitophagie, Apoptose, die Beziehung der Mitochondrien zum endoplasmatischen Retikulum und die Dynamik der Mitochondrien. Membranen zellulärer Organellen bestehen aus Lipiden und Proteinen. Der Umbau dieser Membranen wird durch Wechselwirkungen zwischen spezifischen Lipiden und Proteinen gesteuert.
Gesättigte Fettsäuren umfassen Palmitinsäure (C16) und Stearinsäure (C18). Es wurde gezeigt, dass die Verwendung von Stearinsäure (C18: 0) den Prozess der Mitochondrienfusion stimuliert. Seine Wirkung ist mit einer Wirkung auf Mitofusine verbunden. Bei Mäusen können Nahrungsergänzungsmittel mit Stearinsäure die mitochondriale Dysfunktion, die durch Mutationen in den Pink1- oder Parkin-Genen verursacht wird, teilweise wiederherstellen. Bei Neutrophilen von Menschen, die 2 Tage lang eine C18: 0-arme Diät hatten, befinden sich die Mitochondrien in einem fragmentierten Zustand (50% der Zellen hatten MX fragmentiert, 10% waren mit MX verbunden). Die Verwendung von Stearinsäure führte dazu, dass sie nach 3 Stunden Mitochondrien verschmolzen [8]. Daher ist Sterinsäure wichtig für die Aufrechterhaltung der mitochondrialen Dynamik. Die meiste Stearinsäure kommt in Kakaobohnen vor (31-34%).
Phospholipide sind die Hauptkomponenten von Organellenmembranen. Sie regulieren auch die Dynamik der Mitochondrien und ihre Wirkung ist unterschiedlich [9].
Cardiolipin (CL) stimuliert die mitochondriale Teilung und Fusion innerer Membranen.
Cardiolipin ist für den Betrieb des Komplexes IV (Citrochrom C-Oxidase) der Elektronentransportkette erforderlich. Cardiolipin befindet sich fast ausschließlich in der inneren Membran der Mitochondrien. Mit zunehmendem Alter nimmt die Menge an Cardiolipin ab. Es gibt eine Theorie, dass der Verlust der Cardiolipinfunktion mit dem Ersatz gesättigter Fettsäuren in ihrem Molekül durch mehrfach ungesättigte Fettsäuren verbunden ist. Um dieses Problem zu lösen, müssen gesättigte Fette, die vor allem an Stearinsäure reich sind, in die Ernährung aufgenommen werden.
Um die Effizienz der Abgabe gesättigter Fettsäuren an die Membran zu erhöhen, können Transporter verwendet werden. Zum Beispiel die Verwendung von gesättigtem Phosphatidylcholin (Dipalmitophosphatidylcholin und Dyseroylphosphatidylcholin), das möglicherweise gesättigte FAs direkt an Cardiolipin liefern könnte [10]. Cholin als Träger passiert leicht das Cytosol und gelangt in die Mitochondrien.
Phosphatidsäure (RA) hemmt die mitochondriale Teilung und stimuliert die Fusion externer Membranen (Abb. 7).
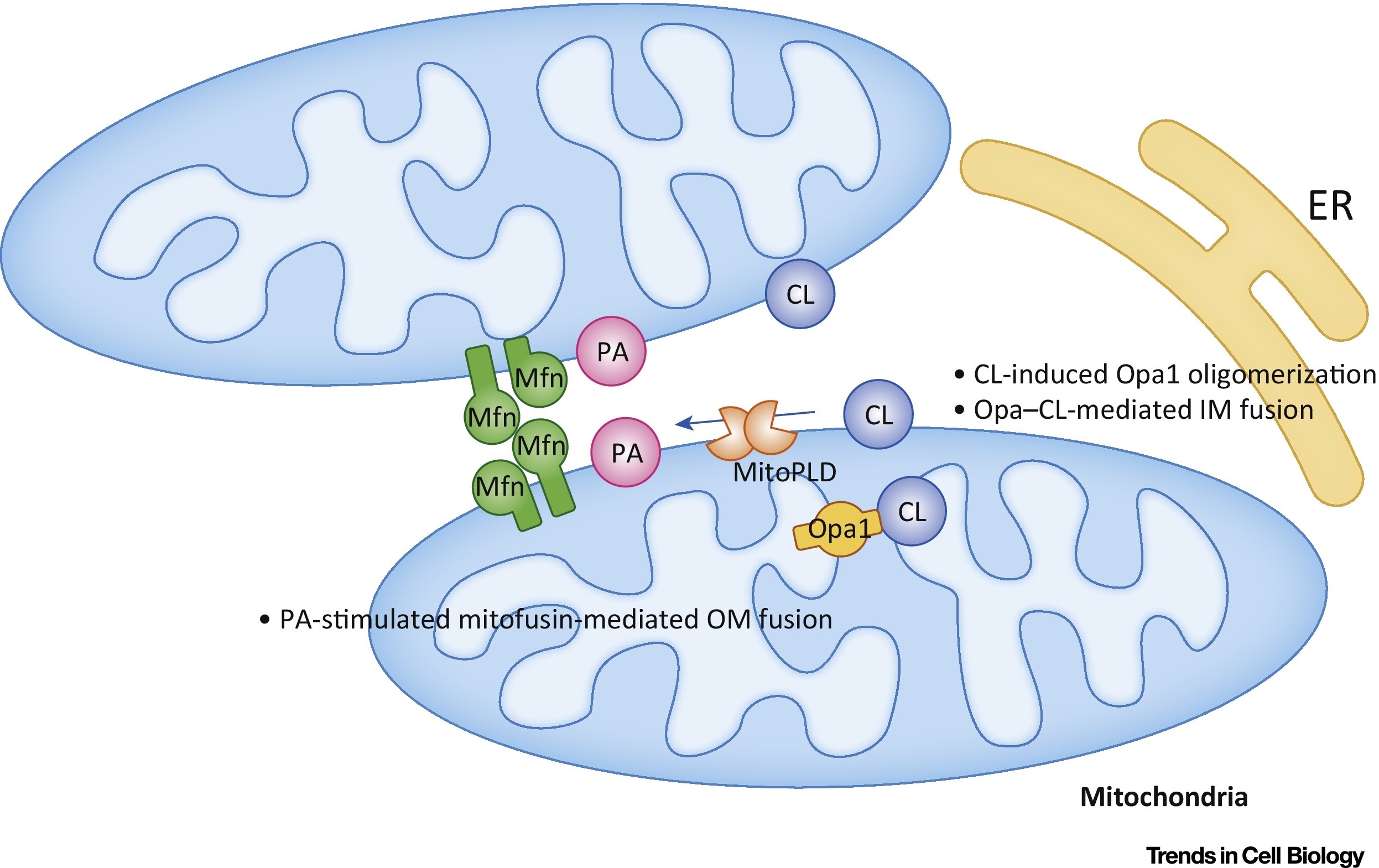 Abb. 7 Regulation der Mitochondrienfusion mit Phosphatidsäure (PA) und Cardiolipin (CL) [von 9].
Abb. 7 Regulation der Mitochondrienfusion mit Phosphatidsäure (PA) und Cardiolipin (CL) [von 9].In der äußeren Membran (OM) stimuliert RA die Mitofusin-vermittelte (Mfn) Fusion. In der inneren Membran (IM) stimuliert CL die Opa1-vermittelte Fusion. Abkürzungen: ER - endoplasmatisches Retikulum; MitoPLD, - Mitochondrien-lokalisierte Phospholipase D.
3. Regulation der Expression von Mitofusinen (Proteine, die für die Dynamik der Mitochondrien verantwortlich sind)Alles, worüber wir oben gesprochen haben (Kalorienrestriktion, Stearinsäure, Phospholipide), beeinflusst die Expression von Mitofusinen.Darüber hinaus gibt es eine Reihe von Medikamenten, die indirekt die Dynamik von Mitochondrien beeinflussen können. Dazu gehört die Verwendung von Metformin.Am interessantesten ist die Verwendung von Substanzen, die die Expression von Mitofusinen direkt beeinflussen können. Eines der potenziellen Medikamente namens Leflunomid (Leflunomid), das von der FDA zugelassen wurde [5,11]. Es ist ein Induktor der Expression von Mfn1 und Mfn2 und wurde als Medikament zur Behandlung von rheumatoider Arthritis registriert.Mitochondriale Gentherapie
Eine beeinträchtigte mitochondriale Dynamik kann mit einer beeinträchtigten Expression von Proteinen verbunden sein, die für die Fusion und Teilung der Mitochondrien verantwortlich sind. Darüber hinaus kann eine Funktionsstörung dieser Proteine mit ihren Mutationen assoziiert sein (und dies geschieht am häufigsten). Es gibt zwei Ansätze zur Berücksichtigung von Ursache-Wirkungs-Wechselwirkungen bei mitochondrialen Dysfunktionen.Es wurde zuvor angenommen, dass Lebensstil, einschließlich übermäßiges Essen, zur Bildung von freien Radikalen, oxidativem Stress, Mutationen des mitochondrialen Genoms und folglich zu Funktionsstörungen der Mitochondrien führt. In jüngster Zeit gibt es jedoch überzeugende Beweise dafür, dass Mutationen mitochondrialer DNA unvermeidlich sind, alle heteroplasmatische DNA-Punktmutationen aufweisen und mit Replikationsfehlern und nicht mit oxidativen Schäden verbunden sind, für die mitochondriale DNA ziemlich stabil ist [12]. Einige unserer Mitochondrien tragen bereits im Stadium des befruchteten Eies Mutationen. Im Laufe der Zeit teilen sie sich, es gibt mehr mutierte Mitochondrien, sie können ihre Funktion nicht normal ausführen.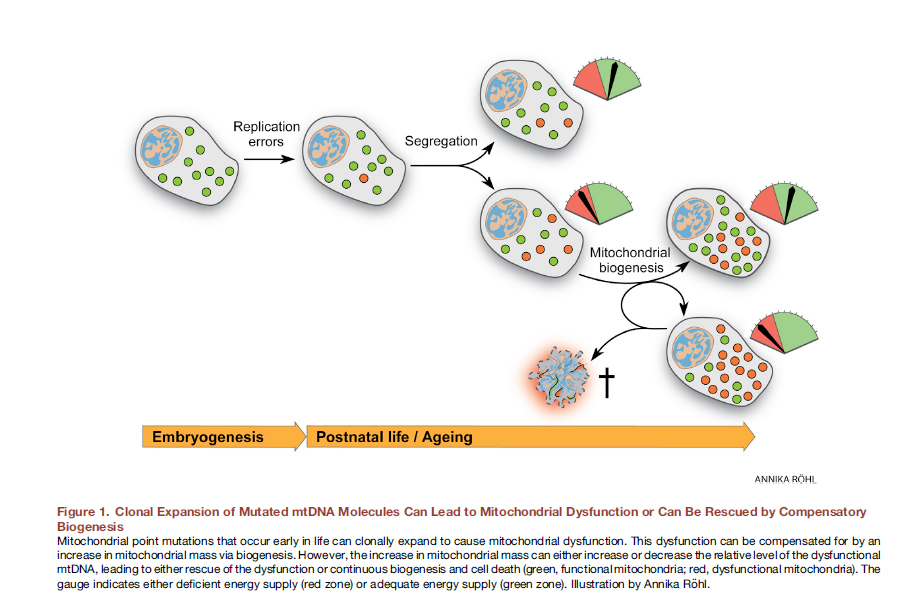 Abb. 8 Die klonale Expansion mutierter mtDNA-Moleküle kann zu einer mitochondrialen Dysfunktion führen oder durch kompensatorische Biogenese "gerettet" werden [von 12].Hier könnte in vivo die Bearbeitung des mitochondrialen Genoms sehr nützlich sein. Es wurde gezeigt, dass für heteroplasmatische DNA-Punktmutationen in Mäusen bereits signifikante Erfolge mit gezielten Zinkfinger-Nukleasen (mtZFN) unter Abgabe eines adenoviralen Vektors erzielt wurden [13].
Abb. 8 Die klonale Expansion mutierter mtDNA-Moleküle kann zu einer mitochondrialen Dysfunktion führen oder durch kompensatorische Biogenese "gerettet" werden [von 12].Hier könnte in vivo die Bearbeitung des mitochondrialen Genoms sehr nützlich sein. Es wurde gezeigt, dass für heteroplasmatische DNA-Punktmutationen in Mäusen bereits signifikante Erfolge mit gezielten Zinkfinger-Nukleasen (mtZFN) unter Abgabe eines adenoviralen Vektors erzielt wurden [13].Mitochondrialer Transfer
Eine weitere vielversprechende Methode zur Beseitigung der mitochondrialen Dysfunktion ist die mitochondriale Transplantation. Die Essenz dieses Ansatzes besteht darin, beschädigte Mitochondrien durch gesunde exogene Mitochondrien zu "ersetzen". Dieser Ansatz wurde erstmals klinisch bei Kindern mit Myokardischämie angewendet. Zur Transplantation wurden autologe isolierte Mitochondrien verwendet, die mit dem Musculus rectus abdominis isoliert wurden (eine Biopsie wurde durchgeführt und dann das Präparat präpariert) und dann durch direkte Injektion verabreicht [14]. Es werden verschiedene Ansätze zur Einführung von Mitochondrien erarbeitet: direkte Injektion isolierter Mitochondrien (lokale Injektion) und systemische Injektion in den Blutkreislauf, wenn die Mitochondrien selbst nach der Zelle „suchen“, in die sie gehen sollen. Forschergruppen untersuchen die Möglichkeit einer mitochondrialen Transplantation bei Parkinson, Leberischämie, Schlaganfall und mitochondrialen Erkrankungen [15].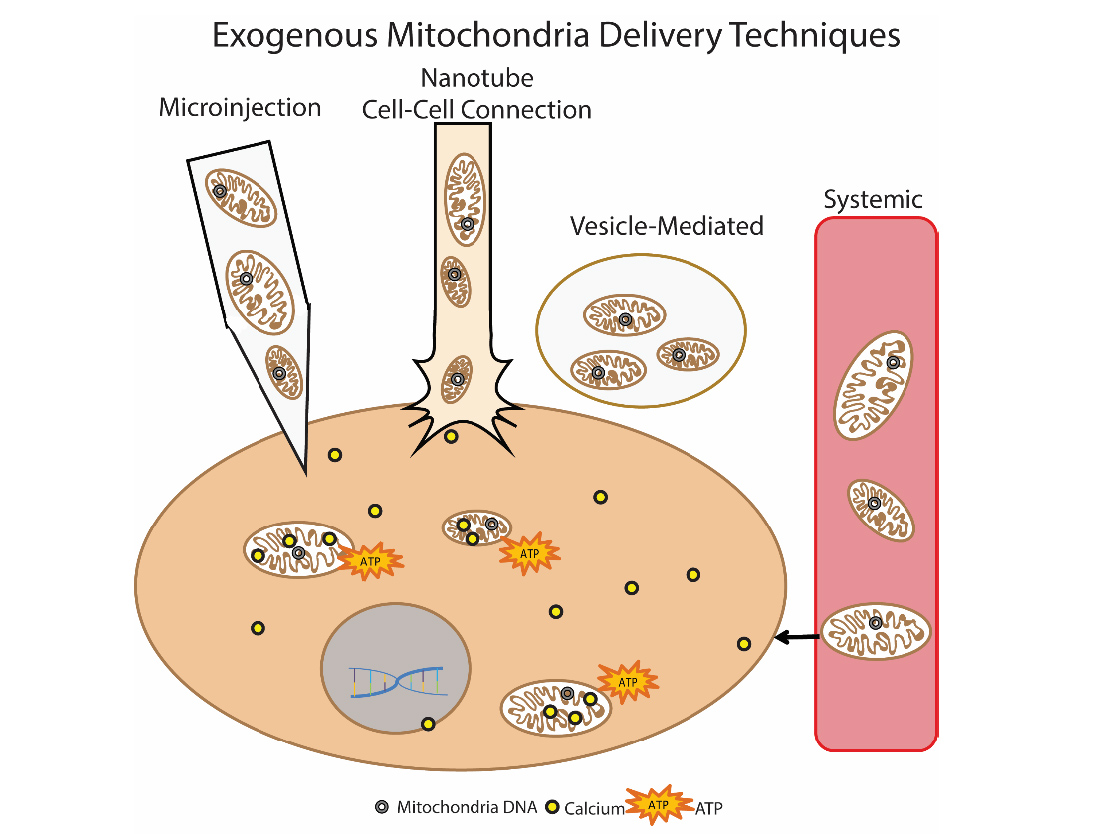 Abb. 9 Methoden zur Abgabe exogener Mitochondrien an die Zelle.Autorin Olga Borisova
Abb. 9 Methoden zur Abgabe exogener Mitochondrien an die Zelle.Autorin Olga BorisovaLiteratur1. Kauppila, Timo ES, Johanna HK Kauppila, and Nils-Göran Larsson. «Mammalian mitochondria and aging: an update.» Cell metabolism 25.1 (2017): 57-71.
www.sciencedirect.com/science/article/pii/S15504131163050222. Schrepfer, Emilie, and Luca Scorrano. «Mitofusins, from mitochondria to metabolism.» Molecular cell 61.5 (2016): 683-694.
www.sciencedirect.com/science/article/pii/S1097276516001337#fig13. Marc Liesa, Orian Shirihai “Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure” Cell methabolism (2013): 491-506
www.sciencedirect.com/science/article/pii/S1550413113001046#fig34. Ramos, Eduardo Silva, Nils-Göran Larsson, and Arnaud Mourier. «Bioenergetic roles of mitochondrial fusion.» Biochimica et Biophysica Acta (BBA)-Bioenergetics 1857.8 (2016): 1277-1283.
www.sciencedirect.com/science/article/pii/S00052728163008585. Cunarro, Juan, et al. «Hypothalamic mitochondrial dysfunction as a target in obesity and metabolic disease.» Frontiers in endocrinology 9 (2018): 283.
www.frontiersin.org/articles/10.3389/fendo.2018.00283/full6. Marcelo O.Dietrich et al. «Mitochondrial Dynamics Controlled by Mitofusins Regulate Agrp Neuronal Activity and Diet-Induced Obesity”.
www.sciencedirect.com/science/article/pii/S0092867413010957#figs27. Steculorum, Sophie M., and Jens C. Brüning. „Sweet mitochondrial dynamics in VMH neurons.“ Cell metabolism 23.4 (2016): 577-579.
www.sciencedirect.com/science/article/pii/S15504131163011768. Senyilmaz-Tiebe, Deniz, et al. „Dietary stearic acid regulates mitochondria in vivo in humans.“ Nature communications 9.1 (2018): 3129.
www.nature.com/articles/s41467-018-05614-69. Kameoka, Shoichiro, et al. „Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics.“ Trends in cell biology (2017).
www.sciencedirect.com/science/article/pii/S096289241730158710.
raypeatforum.com/community/threads/mitolipin-liquid-saturated-phosphatidylcholine-pc-mix.1039811. Miret-Casals, Laia, et al. „Identification of new activators of mitochondrial fusion reveals a link between mitochondrial morphology and pyrimidine metabolism.“ Cell chemical biology25.3 (2018): 268-278.
12. Kauppila, Timo ES, Johanna HK Kauppila, and Nils-Göran Larsson. „Mammalian mitochondria and aging: an update.“ Cell metabolism 25.1 (2017): 57-71.
13. Gammage et al. “Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo” Nature medicine, 2017
www.nature.com/articles/s41591-018-0165-914. Emani, Sitaram M., et al. „Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury.“ The Journal of thoracic and cardiovascular surgery 154.1 (2017): 286-289.
www.jtcvs.org/article/S0022-5223 (17)30258-1/fulltext
15. McCully, James D., et al. „Mitochondrial transplantation: From animal models to clinical use in humans.“ Mitochondrion 34 (2017): 127-134.
www.sciencedirect.com/science/article/pii/S1567724917300053