Einführung
Worum geht es in diesem Text?
Wenn eine Person von einer "Simulation der Realität" hört, wird sie höchstwahrscheinlich verschiedene Science-Fiction-Werke (wie Matrix, The Dark City oder Zero Theorems) oder Computerspiele entwickeln. Bei Personen, deren Köpfe durch einen Ingenieurabschluss verstopft sind, werden
möglicherweise Pakete wie
KOMPAS-3D AutoCAD, Solid Edge oder NX angezeigt. Eine Person, die der Wissenschaft zuhört, wird sich wahrscheinlich an jede
Modellierung verschiedener Weltraumgeräte erinnern.
Aber es gibt noch eine weitere Ebene der Realität, die sich als unverdient vergessen herausstellen wird: Die Ebene, auf der die gesamte Chemie stattfindet, ist die Ebene der Atome und Moleküle. Es kann auch sehr erfolgreich auf einem Computer simuliert werden. Da die Quantenmechanik in diesem Abschnitt der Realität für alles verantwortlich ist, werden solche Berechnungen oft als Quantenchemie bezeichnet. Und wir werden über seinen Zusammenhang mit der Realität sprechen, die mit experimentellen Methoden untersucht wurde.
In diesem Text geht es um die elementarsten Dinge. Die Praxis, wissenschaftliche Zeitschriften zu lesen und verschiedene Berichte anzuhören, zeigt jedoch, dass dies ständig daran erinnert werden sollte.
Der Text richtet sich an Personen, die verstehen und / oder daran interessiert sind, wie Atome und Moleküle leben.Entnommen aus xkcd.comKurzer HintergrundSo kam es, dass ein Stipendiat, der leider in der russischen Wissenschaft arbeitete, mich einlud, einen Vortrag in seinem Spezialkurs für 2 Personen an einer der bekanntesten physischen Universitäten Russlands zu halten. Aber durch einen seltsamen Zufall wurde sie zu einer parallel abgehaltenen Studentenkonferenz versetzt ... Dort erregte sie bei den Studenten kein großes Interesse, und das Material tat mir sehr, sehr leid. Deshalb beschloss ich, Habr ein wenig zu spammen und zu versuchen, die pädagogische Vorlesung in einen populärwissenschaftlichen Artikel zu verwandeln.
Physikalische Methoden zur Untersuchung des Lebens von Molekülen
Aus Schulchemie- und Physikkursen wissen wir, dass alle Substanzen aus Atomen, Molekülen, Ionen oder Kombinationen davon bestehen. Und wir scheinen sogar zu wissen, welche Art von Leben sie leben. Aber diese Informationen sollten ihre eigenen zuverlässigen Quellen (Forschungsmethoden) haben, und das sind sie wirklich.
Es gibt viele, viele Möglichkeiten, das Leben von Atomen auszuspionieren. Wer dies wünscht, kann sich beispielsweise in klassischen Lehrbüchern mit einigen von ihnen genauer vertraut machen
- Pentin Yu.A., Vilkov L.V. Physikalische Forschungsmethoden in der Chemie. - M.: Mir, 2006,
- Drago R. Physikalische Methoden in der Chemie. - M.: Mir, 1981.
Aber grob und ziemlich leicht fallen drei Hauptgruppen von Methoden auf:
- spektroskopische Methoden
- Beugungsmethoden
- verschiedene Methoden der Mikroskopie (egal, durchscheinend oder scannend, für uns ist dies jetzt nicht unbedingt erforderlich).
Über Letzteres wird nicht gesprochen, aber seine Werkzeuge sind nicht weniger wichtig als die ersten beiden.
Warum wird nicht über Mikroskopie gesprochen?(Ich scheue mich in der Mikroskopie überhaupt nicht vor dem Wort)
Spektroskopische Methoden zur Untersuchung von Materie
Diese mächtige Gruppe von Methoden bietet uns sehr, sehr viele Dinge: von der Suche und Bestimmung von Molekülen im interstellaren Medium und auf anderen Planeten bis zur banalen Überprüfung auf Sprengstoff am Flughafen.
Das allgemeine Prinzip der Spektralmethoden
Wenn von Spektroskopie die Rede ist, wird normalerweise auf das folgende allgemeine Funktionsprinzip Bezug genommen.
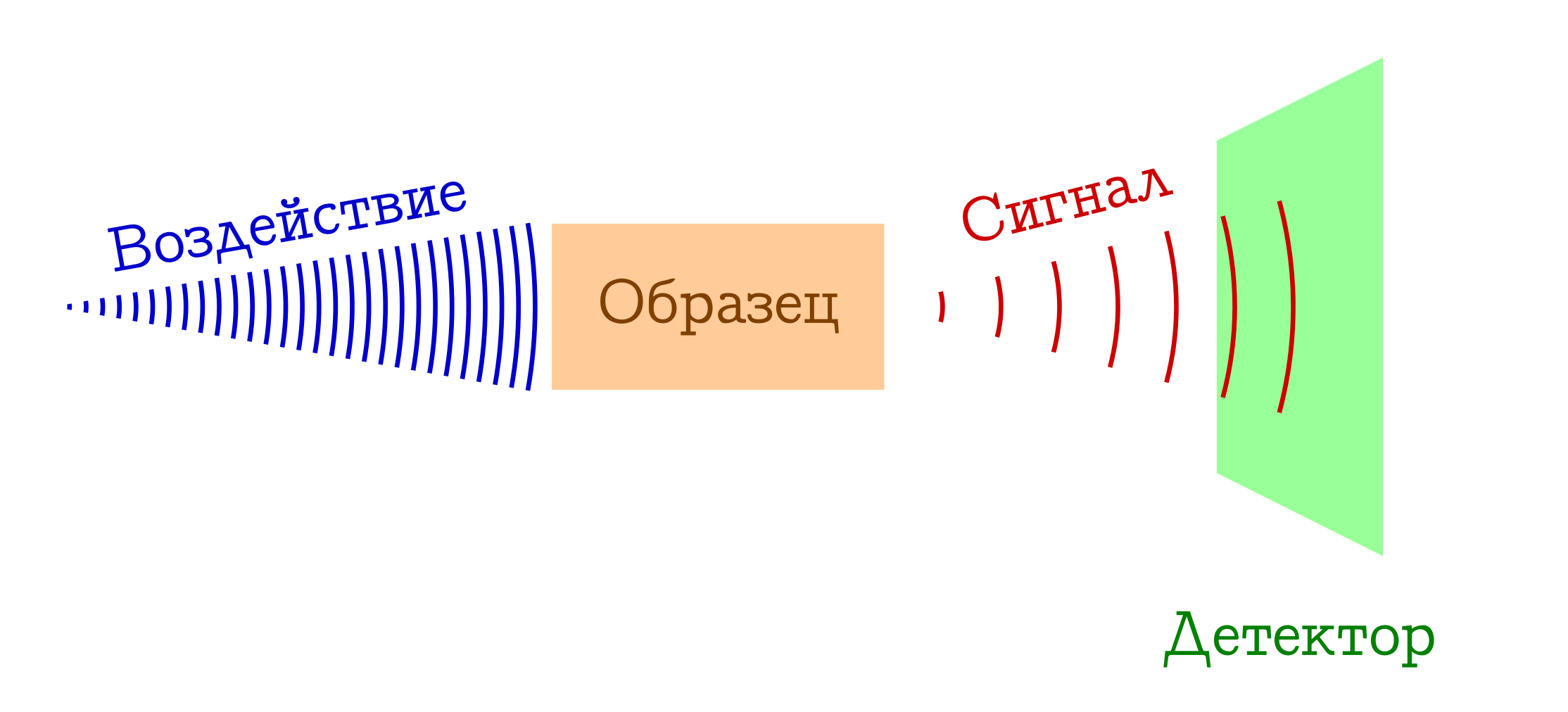
Das allgemeine Schema spektraler Methoden zur Untersuchung von Substanzen
- Wir haben etwas, mit dem wir (zum Beispiel eine Glühbirne / Laser / Sonnenlicht) auf die für uns interessante Probe einwirken. Meistens handelt es sich um eine elektromagnetische Studie, aber es können auch Elektronen sein (zum Beispiel in der Massenspektroskopie mit Ionisation durch Elektronenstoß) oder ein Cocktail von allem, was aus Plasma möglich und unmöglich ist (zum Beispiel in der Flammenspektroskopie , die von Schulkindern und Studenten der chemischen Fakultät so geliebt wird). Auf die eine oder andere Weise muss etwas an unserer Probe funktionieren.
- Wenn es einer Probe ausgesetzt wird, passiert etwas, das seinen Zustand ändert. Dies kann ein Übergang zu einer Art angeregtem Niveau (bei jeder Spektrophotometrie oder Raman-Spektroskopie) oder sogar der Zusammenbruch des molekularen Systems (wie bei Massenspektren oder Photoelektronenspektroskopie ) sein. Aber irgendwie sollte das Muster irgendwann anders sein.
- ???
- GEWINN !!! Wir registrieren ein bestimmtes Signal (emittiert oder absorbiert) mit dieser Änderung in der Probe auf molekularer Ebene. Hierbei können Photonen verloren gehen, die für den Wechsel der Probe aufgewendet wurden (dann haben wir Absorptionsspektroskopie), oder umgekehrt, überschüssige Photonen, die nach vorläufiger Anregung der Substanz emittiert werden (Emissionsspektroskopie), eine Änderung der Wellenlänge der anfänglichen Photonen infolge der Wechselwirkung mit der Substanz (Raman-Spektroskopie, mehr) im Ausland als
Ramenovskaya Ramanova bekannt ) oder dumm Fragmente der ursprünglichen Moleküle (wie in Massenspektren oder Photoelektronenspektroskopie ). Es gibt viele Möglichkeiten - eine Essenz: Es gibt ein Signal!
Ein Beispiel für solche Methoden sind verschiedene Buchstaben: NMR, ESR, MW, THz, IR, UV / Vis, RFA, MS, PES, EXAFS, XANES usw. usw.
Alle (oder viele von ihnen) sind jedem Chemiker vertraut (oder sollten vertraut sein). Alle diese Methoden sind das (alles andere als unvollständige) Standardarsenal eines sich selbst respektierenden Forschers, der sich mit Substanzen befasst.
Spektralbereiche und ihre Beziehung zum Leben von Molekülen
 Entnommen aus xkcd.com
Entnommen aus xkcd.comDa die Spektroskopie in den allermeisten Fällen immer noch an elektromagnetische Strahlung gebunden ist, ist es logisch, die Bereiche des elektromagnetischen Spektrums mit verschiedenen Aspekten des atomar-molekularen Lebens zu verknüpfen. Schließlich ist die Frequenz der in der Spektroskopie verwendeten elektromagnetischen Wellen eine Art „Uhr“, mit der Sie feststellen können, wie lange dieser oder jener Prozess in molekularen Systemen dauert. Wenn Sie diese Frequenz ändern, können Sie verschiedene molekulare Prozesse untersuchen (und sogar darauf reagieren).
Also.
- Aus chemischer Sicht passiert im superlangen Wellenlängenbereich nichts Interessantes, so dass Sie sich nicht daran erinnern können.
- Mit der Frequenz von Radio und Mikrowellen (und sogar langwelliger Infrarotstrahlung, IR = IR) drehen sich verschiedene Moleküle in der Gasphase: groß und schwer - im Bereich der Radiowellen (niedrigere Frequenzen) und klein und leicht - im IR (höhere Frequenzen).
- Im IR treten jedoch (hauptsächlich) verschiedene molekulare Schwingungen auf: Alle Konformations- und anderen nicht offensichtlichen Bewegungen innerhalb der Moleküle finden im langwelligen IR statt, und Streckschwingungen (Dehnung - Verkürzung der chemischen Bindungslängen) treten im kurzwelligen Bereich (bis zu 4000 cm –1 ) auf.
- Nun, dann kommt der Ort des Spektrums, an dem verschiedene elektronische Übergänge leben (bis zur Region der γ-Quanten). Bei niedrigeren Frequenzen (sichtbar, UV = UV und weiches Röntgen) leben hauptsächlich Übergänge, die mit Valenzelektronen verbunden sind.
Warum sehen wir?Übrigens können wir gerade aufgrund elektronischer Übergänge sehen: In unseren Augen (in Zapfen) gibt es Strukturen, deren Zusammensetzung eine
Netzhaut aufweist . Wenn ein sichtbares Photon von diesem Molekül absorbiert wird, bricht eine Doppelbindung darin, was zur cis-trans-Isomerisierung führt. Und genau diese Veränderung nehmen wir als primäres Signal wahr, das dann an unser Gehirn übertragen wird.
Aber mit zunehmender Photonenenergie (d. H. Mit zunehmender Frequenz, wie wir uns aus der Planck-Formel erinnern E = h n u ) Wir gelangen zu immer tieferen Schichten der elektronischen Struktur, bis wir im Röntgenbereich bis zu den letzten 1s-Schalen ( oder, wie sie Röntgen genannt werden, K ) ruhen.
Wenn wir also die richtige Wellenlänge der elektromagnetischen Strahlung wählen, können wir einen bestimmten Prozess in den Molekülen genauer betrachten.
Substanzbeugungsmethoden
Lassen Sie uns nun ein wenig über Beugung sprechen. Das schematische Diagramm solcher Experimente ist ebenfalls einfach.
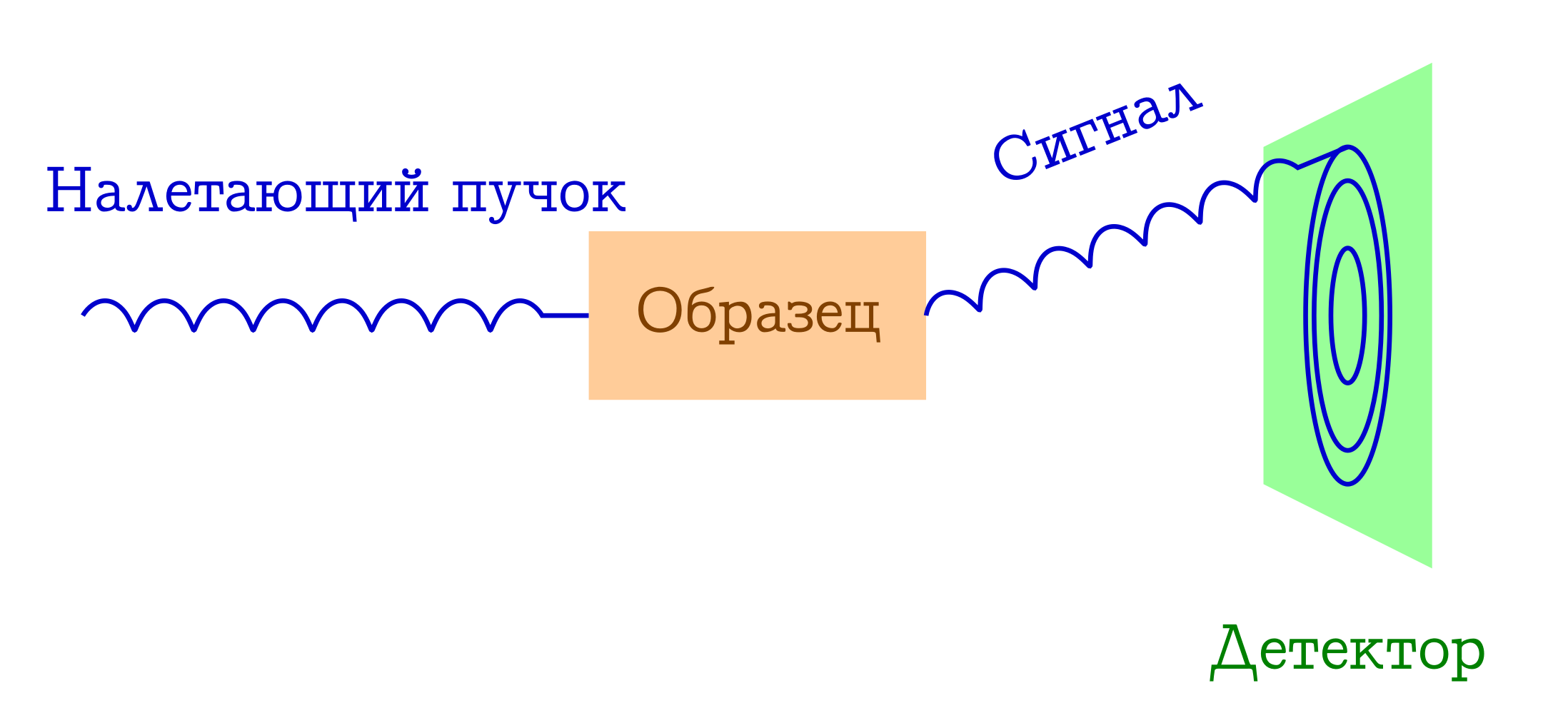
Allgemeines Schema der Beugungsmethoden zur Untersuchung von Substanzen
- Ein Strahl einiger Partikel fliegt auf die Probe. Meistens sind es entweder Röntgenphotonen oder Elektronen oder Neutronen.
- Diese Teilchen streuen durch verschiedene Mechanismen elastisch an Atomen in der für uns interessanten Probe (d. H. Ohne die Wellenlänge und Phase der Welle zu ändern, ändern sie einfach die Flugrichtung). Mit der Probe dieser einfallenden Partikel passiert nichts: Sie hat einfach keine Zeit, darauf zu reagieren.
- Die interatomaren Abstände dienen als Beugungsgitter für den einfallenden Strahl, daher sehen wir ein schönes Beugungsbild auf dem Detektor.
Aus dem letzten Absatz ergibt sich die Bedingung für die Wellenlänge einfallender Teilchen (λ): Sie sollte in der gleichen Größenordnung oder kleiner als die charakteristische Ordnung der interatomaren Abstände sein, so dass das typische λ für diese Methoden 1 - 0,01 Å beträgt.
Die Hauptfehlerarten beim Vergleich von Experimenten und theoretischen Berechnungen
Als Ergebnis haben wir ein sehr interessantes Bild: In der Spektroskopie und in der Beugung beobachten wir eine Art linkes Signal, das irgendwie
indirekt anzeigt, was tatsächlich im molekularen System passiert.
Die Analogie zur platonischen HöhleDieses Gemälde erinnert unheimlich an den
Mythos der Platonhöhle . Wir haben eine bestimmte reale Welt der Moleküle. Aber wir sehen nur Schatten von ihm an der Höhlenwand (Detektor), die eine unvollständige Darstellung aller interessanten Dinge sind, die auf dieser Ebene der Realität geschehen.
Glücklicherweise können wir manchmal theoretisch das für uns interessierende Signal berechnen (wie zum Beispiel in der Mikrowellen-, IR- oder UV / Vis-Spektroskopie), und manchmal können wir aus dem beobachteten Signal die interessierenden Größen extrahieren, die für die quantenchemische Berechnung verfügbar sind (zum Beispiel Abstand zwischen Atomen in einem Molekül, Dipolmoment usw.). Und hier haben wir die Chance, dass sich das numerische und das reale Experiment in der leidenschaftlichen Phase des Vergleichs miteinander vereinen können ... und hier können standardmäßig 4 Arten von Fehlern auftreten.
Achtung! Der Begriff „Fehler“ bedeutet hier nicht, dass das Ergebnis des Vergleichs offensichtlich falsch ist. Es ist nur so, dass der Vergleichsgrund sehr wackelig und sumpfig wird und ein schlampiger Schritt die ganze Arbeit leicht durcheinander bringen kann.
- Unterschiedliche Bedingungen des Experiments und / oder der Berechnung (Aggregationszustand, Temperatur, Druck usw.). Wir können plötzlich anfangen, verschiedene Systeme untereinander zu vergleichen, aus irgendeinem Grund, wenn wir sie als gleich betrachten. Zum Beispiel ist es offensichtlich, dass das Hinzufügen von einem oder fünf Teelöffeln Zucker zu einer Tasse Tee zu demselben physikalischen System führt, das als „Tee mit Zucker“ bezeichnet wird, aber die Eigenschaften dieses Systems sind sehr unterschiedlich. Und es kann leicht gemessen werden. Zum Beispiel mit einem Thermometer (Messung der Temperatur des Tees unmittelbar nach dem Auflösen des Zuckers) oder mit der Zunge (eine der sogenannten organoleptischen Analysemethoden). Wenn wir also die resultierenden Systeme miteinander vergleichen (sei es eine echte Tasse Zucker mit Tee oder sein Computermodell), dürfen wir nicht vergessen, dass die Ähnlichkeiten ihre Grenzen haben und dass wir, wenn wir die Fehlerquote für „Ähnlichkeit“ verringern, schließlich die Unterschiede finden werden.
- Unterschiedliche physikalische und / oder mathematische Bedeutung der Parameter (der Parameter der physikalischen Bedeutung im üblichen Sinne existiert möglicherweise nicht einmal). Auch hier ist alles einfach: Wenn wir 2 Größen mit einem ähnlichen Namen vergleichen, bedeutet dies nicht, dass die Größen dieselbe physikalische Bedeutung haben. Zum Beispiel die stellvertretende Bewertung unter der Gesamtbevölkerung der Stadt vs. Bewertung nur unter Omas. Sowohl diese als auch diese Bewertung (was auch immer es ist), diese Zahlen (oder was auch immer es ist) können sogar stark miteinander korrelieren, aber die Bedeutung dieser Parameter ist immer noch unterschiedlich, und dieser Unterschied kann erkannt werden.
- "Zufällige" Fehler . Dies schließt einige systematische Fehler ein, die dem Experimentator / Simulatortheoretiker nicht bekannt sind, oder wirklich zufällige Fehler im Experiment / in der Berechnung, die nicht kontrolliert und / oder vorhergesagt werden können. Im Prinzip können solche Dinge selbst Gegenstand der Untersuchung verschiedener interessanter systematischer Effekte werden.
oder nur eine Schätzung des nützlichsten S / N-Verhältnisses („Signal zu Rauschen“). - Und der letzte Standardfehler ist das Wachstum der Hände des Experimentators / Rechners aus dem Beckenknochen, dh gewöhnliche menschliche Fehler. Es ist nicht erforderlich, irgendetwas zu untersuchen. Überprüfen Sie einfach die Arbeit oder wiederholen Sie das Experiment, um den entsprechenden Pfosten zu finden und zu beseitigen.
Über die letzten beiden Arten von Fehlern kann nichts Konkreteres gesagt werden, aber über die ersten beiden, und wenn Sie eine bestimmte Forschungsmethode anwenden, können Sie viele Dinge sagen. Deshalb werden wir uns auf sie konzentrieren. Das Hauptaugenmerk wird in diesem Fall auf den strukturellen Unterschieden der Moleküle liegen.
Fehler Nr. 1. Unterschiede in den molekularen Eigenschaften unter verschiedenen Bedingungen
NaCl: wenn keine Fehler vorliegen
Aus irgendeinem Grund fällt niemandem ein, dass der Einkristall aus Natriumchlorid (NaCl), einem riesigen Molekül aus Na
+ - und Cl
- -Ionen, und das zweiatomige NaCl-Molekül, das durch Verdampfung dieses Kristalls bei verrückten Temperaturen erhalten wird, ein und dasselbe haben Sagen wir die Struktur.
Und selbst wenn wir davon ausgehen, dass zumindest die Abstände zwischen Chlor und Natrium (
r NaCl ) hier und da gleich sind, wird uns das Experiment in die richtige Position bringen:
Wo machen wir einen Fehler?Tatsächlich erlauben wir bei einem solchen Vergleich die Möglichkeit von Fehler Nr. 2, aber hier ist alles in Ordnung. Wenn wir die Fehler eines solchen Vergleichs bewerten, liegen sie in der Größenordnung von 0,01 Å, was erheblich weniger ist als die Differenz der verglichenen Parameter. Das heißt, Dies ist kein Fehler, sondern ein echter Effekt.
Wie Sie den Abstand zwischen dem Natriumkation und dem Chloranion in einem Salzkristall selbst ermitteln könnenDas Ermitteln des Abstands zwischen Atomen in einem zweiatomigen NaCl-Molekül aus experimentellen Daten ist ebenfalls kein so kompliziertes Verfahren. Das Problem ist aber nur, dass ein solches Experiment eine komplizierte Sache ist. Daher ist es einfacher, eine Datenbank zu verwenden, in der die erforderlichen Entfernungen bereits angegeben sind.
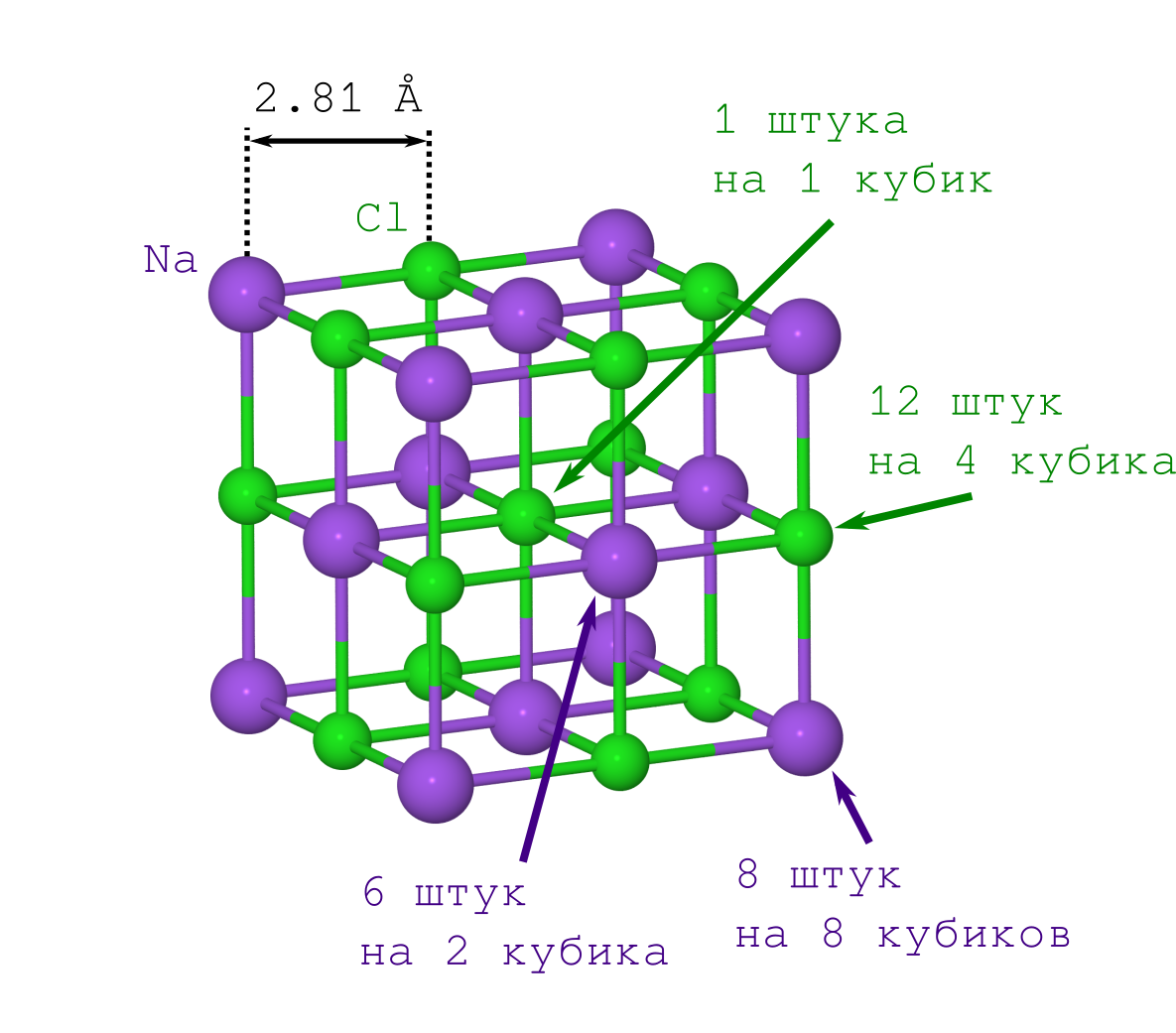
Um jedoch den Abstand zwischen den Atomen im Kristall zu erhalten, reicht nur die Dichte des kristallinen Salzes der Tabelle ρ = 2,165 g / cm
3 aus, die
leicht von Wikipedia bezogen und selbst zu Hause gemessen werden kann.
Um die Entfernung zu berechnen, benötigen wir:
- die Dichte des NaCl-Kristalls (ist),
- Kenntnis der Position der Ionen dieses Kristalls.
Wenn Sie dies zum ersten Mal getan hätten (etwa zu Beginn des 20. Jahrhunderts), müssten Sie sich mit dem zweiten Punkt quälen. Dies ist den modernen Menschen jedoch bereits bekannt: Das NaCl-Gitter hat die Form eines Würfels, in dem sich Na
+ - und Cl
- -Ionen abwechseln (siehe Bild oben). Durch Multiplizieren des angegebenen Kristallfragments („Kopieren-Einfügen“ des angegebenen Teils und Anpassen an die vorherige Iteration von Angesicht zu Angesicht) erhalten wir einen NaCl-Kristall mit jeder gewünschten Größe und jeder gewünschten (Minecraft-) Form.
Die Dichte dieses Würfels sollte also der des gesamten Kristalls entsprechen. Angesichts dieser Dichte ist
rho= fracmV (d. h. Masse pro Volumen), es stellt sich heraus, dass wir den Abstand zwischen den Atomen berechnen können, wenn wir die Masse und den geometrischen Ausdruck für das Volumen kennen.
Das Volumen des Würfels ist offensichtlich: Die Länge der Rippe ist doppelt so groß wie der Abstand Na - Cl (
L=2r mathrmNaCl ), was bedeutet, dass das gewünschte Volumen ist
V=L3=8r mathrmNaCl3 .
Die Masse ist nicht so einfach. Die meisten unserer Atome liegen auf den Eckpunkten, Kanten und Flächen des Würfels, was bedeutet, dass sie gleichzeitig zu mehreren dieser Würfel gehören. Dies muss bei den Berechnungen berücksichtigt werden.
Beginnen wir mit Na
+ -Ionen. Wir haben nur 2 Arten von ihnen (siehe das Kristallgittermuster):
- diejenigen, die sich an den Eckpunkten des Würfels befinden (es gibt so viele wie die Eckpunkte des Würfels, d. h. 8, und sie befinden sich gleichzeitig in 8 Würfeln, sodass Sie diese Zahl durch 8 teilen müssen),
- diejenigen, die auf den Gesichtern liegen (es gibt 6 von ihnen, und sie gehören gleichzeitig zu 2 Würfeln).
Als Ergebnis erhalten wir, dass unser Würfel enthält
8 cdot frac18+6 cdot frac12=4 Natriumion.
Nun zu Cl
- . Es gibt auch nur 2 Arten von ihnen (siehe das Kristallgittermuster):
- diejenigen, die an den Rändern des Würfels liegen (es gibt 12 von ihnen, und sie sind gemeinsam im Besitz von 4 Würfeln),
- das Cl - das ist in der Mitte des Würfels, es ist eins und gehört nur zu unserem Würfel.
Daher enthält unser Würfel
12 cdot frac14+1 cdot frac11=4 Chlorion.
Die Zusammensetzung des Kristalls entspricht offensichtlich der chemischen Formel von NaCl, aber die Masse unseres Würfels ist gleich (vergessen Sie nicht, dass die Atommassen im Periodensystem in
Atommasseneinheiten angegeben sind):
m=4 cdot( underbraceM mathrmNa23 textam+ underbraceM mathrmCl35.5 textamu)=234 textamu=234 cdot1.66 cdot10−24 textr=3.88 cdot10−22 textg\.
Nun aus der Beziehung
rho= fracmV wir können eine Gleichung für die Länge machen
r mathrmNaCl ::
r mathrmNaCl3= left( fracm8 rho right) ,
was leicht zu lösen ist:
r mathrmNaCl= left( fracm8 rho right)1/3= left( frac3.88 cdot10−22 [ textg]8 cdot2.17 [ textg/ textcm3] right)1/3=2.82 cdot10−8 [ textcm]=2.82 [ textÅ] .
Aus den Daten der Röntgenkristallographie von 2,81 Å (z. B. von
Abrahams, SC; Bernstein, JL Genauigkeit eines automatischen Diffraktometers. Messung der Natriumchloridstrukturfaktoren // Acta Crystallographica (1965) 18, 926-932 ) haben wir nur 0,01 Å verpasst, Das ist cool genug.
Jemand könnte denken, dass der Unterschied von 0,45 Å unbedeutend ist, aber dies ist fast der Bohr-Radius (0,52 Å), der dem wahrscheinlichsten Abstand des Elektrons entspricht, und der Unterschied ist nach atomaren Maßstäben enorm.
Warum sich NaCl in Form eines 2-Atommoleküls von einem Kristall unterscheidetHier ist alles sehr einfach. Das unendliche Kristallgitter schafft die Möglichkeit eines irreversiblen "Springens" für 3s
1 Natriumelektronen pro Chloratom, da die resultierende Ladungsdifferenz durch Wechselwirkung mit Nachbarn kompensiert wird.
3s
1 ( ), ,
«» :
Na:Cl↔Na+Cl−
() (), .
,
±1 , , .
NaCl (2.36 Å),
d=q⋅rNaCl=9.0 [] wo
q≥0 (
+q ,
−q )
, « » 0.21, ..
d=0.21⋅9.0=1.9 [qe⋅Å] , :
q=drNaCl=1.92.36=0.8 . «» 0.2 NaCl NaCl .
Ferrocen
Es lohnt sich, von ionischen Kristallen zu molekularen zu wechseln, in denen Moleküle dicht gepackt sind, so dass ein Vergleich plötzlich und ohne Vorbehalte möglich wird.Der Unterschied darf aber nicht vergessen werden. Und es gibt sogar ein klassisches Beispiel zu diesem Thema: ein Ferrocenmolekül .Dies ist die einfachste Sandwichverbindung. Darin befindet sich ein neutrales Eisenatom (wie ein Schnitzel) zwischen zwei fünfgliedrigen aromatischen Ringen (Brötchen).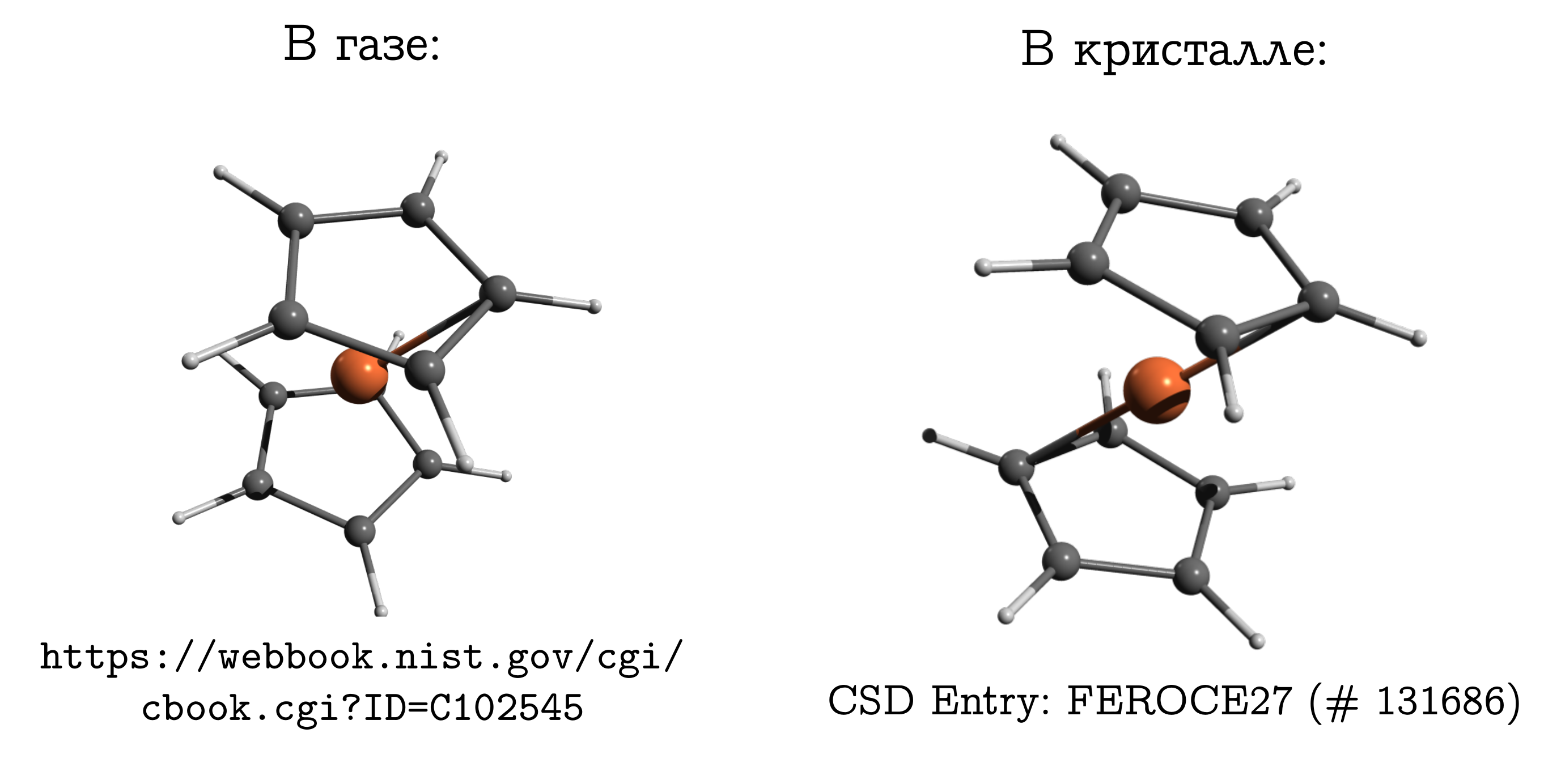 Dieses Molekül kann ziemlich leicht verdampft werden und stellt fest, dass die sogenannte stabilste Struktur in der Gasphase ist behinderte Konformation. Darin liegen sich die Kohlenstoffe und Wasserstoffatome des oberen und unteren Rings gegenüber (siehe Bild oben), da in diesem Fall die Dispersionswechselwirkungen am stärksten sind zwischen diesen Teilen des Moleküls und Dispersion ist immer vorteilhaft.Wenn wir einen Ferrocenkristall nehmen, stellt sich heraus, dass die Moleküle dort eine andere stabile Konformation haben (die für Kohlenwasserstoffe als inhibiert bezeichnet wird), bei der sich Wasserstoff und Kohlenstoff eines Rings über / unter der CC-Bindung des anderen befinden. Es gibt Dispersionswechselwirkungen zwischen Molekülen, und eine ähnliche, scheinbar unbequeme für eine Molekülstruktur ergibt sich aus der Tatsache, dass es für Moleküle einfacher ist, nur in einer unangenehmen Form zusammen zu passen, und diese persönliche Unannehmlichkeit wird durch Wechselwirkung miteinander kompensiert.
Dieses Molekül kann ziemlich leicht verdampft werden und stellt fest, dass die sogenannte stabilste Struktur in der Gasphase ist behinderte Konformation. Darin liegen sich die Kohlenstoffe und Wasserstoffatome des oberen und unteren Rings gegenüber (siehe Bild oben), da in diesem Fall die Dispersionswechselwirkungen am stärksten sind zwischen diesen Teilen des Moleküls und Dispersion ist immer vorteilhaft.Wenn wir einen Ferrocenkristall nehmen, stellt sich heraus, dass die Moleküle dort eine andere stabile Konformation haben (die für Kohlenwasserstoffe als inhibiert bezeichnet wird), bei der sich Wasserstoff und Kohlenstoff eines Rings über / unter der CC-Bindung des anderen befinden. Es gibt Dispersionswechselwirkungen zwischen Molekülen, und eine ähnliche, scheinbar unbequeme für eine Molekülstruktur ergibt sich aus der Tatsache, dass es für Moleküle einfacher ist, nur in einer unangenehmen Form zusammen zu passen, und diese persönliche Unannehmlichkeit wird durch Wechselwirkung miteinander kompensiert.Warum Ferrocen so anders ist als EthanEine Person, die mit Chemie vertraut ist, muss sich normalerweise selbst überwältigen, um sich an die Struktur von Ferrocen in einem Gas zu erinnern. Immerhin hat er Erinnerungen an Ethan (C
2 H
6 ), in dem die stabilste Konformation gehemmt ist (wenn die Wasserstoffatome eines CH
3 -Stücks „zwischen“ den Wasserstoffatomen eines anderen CH
3 liegen ), weil In dieser Position wird die interatomare Abstoßung zwischen den Elektronenschalen von Wasserstoff minimiert.

Adaptiert von
www.chem.msu.su/rus/teaching/stereoUnd hier liegt der ganze Unterschied in der Ferne. Die Standardform des Potentials von Dispersionswechselwirkungen ist das Lennard-Jones-Potential (dies ist übrigens ein, nicht zwei Männer):
V mathrmLJ(r)= fracAr12− fracBr6
Darin wird der erste Term der interatomaren Abstoßung und der zweite der interatomaren Anziehung entnommen, die sich aus Schwankungen der Elektronendichte ergibt. Im Allgemeinen sieht dieses Potenzial ungefähr so aus:
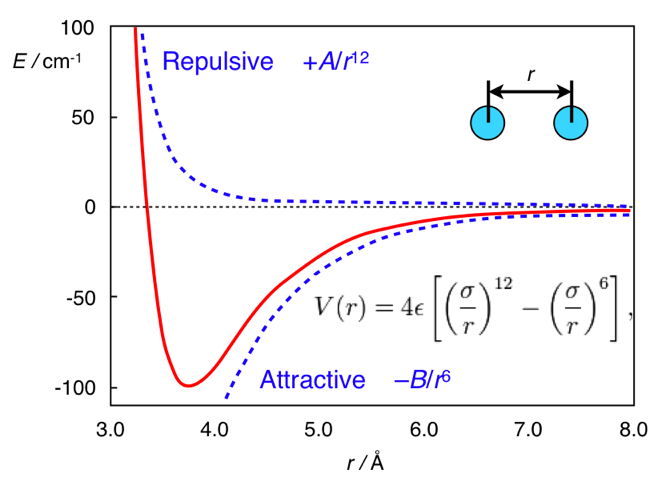
Lennard-Jones-Potenzial. Adaptiert von
Chemistry.stackexchange.com/questions/34214/physical-significance-of-double-well-potential-in-quantum-bondingUnd im Fall von Ethan sind die Wasserstoffatome zu nahe beieinander, so dass sie sich (relativ zu ihrem Minimum) auf der linken Seite der Kurve befinden und durch Abstoßung gekennzeichnet sind. Im Falle von Ferrocen befindet sich zwischen den Ringen eine Schicht von nicht kranker Größe (ein Eisenatom), aufgrund derer die Ringe weit genug sind, um keine interatomare Abstoßung zu fühlen. Und so sind sie im rechten (attraktiven) Teil des Potenzials.
Histamin
Im Falle von Ferrocen haben wir das sogenannte gesehen Konformationsunterschiede: Das Molekül blieb gleich (d. h. es wurden keine chemischen Bindungen aufgebrochen oder gebildet), und seine Form änderte sich geringfügig.
Aber die Unterschiede können zum Beispiel noch größer sein, wenn die sogenannten
tautomere Transformationen . Die Tautomerisierung ist eine Klasse chemischer Reaktionen, die so leicht und schnell ablaufen, dass wir gleichzeitig mehrere Isomere eines Moleküls haben können, die leicht ineinander übergehen. Diese Isomere werden Tautomere genannt.
Ein Standardbeispiel hierfür: Keto-Enol-Tautomerie in Ketonen:

Wie in diesem Beispiel ist Tautomerie meistens mit dem Hüpfen eines Protons von einem warmen Ort zum anderen verbunden. Und diese Reaktionen hängen mit dem
Tunneleffekt zusammen , für den Wasserstoff als leichtestes Atom am anfälligsten ist.
Solche chemischen Umwandlungen sind charakteristisch für viele biologische Moleküle, beispielsweise die
stickstoffhaltigen Basen, aus denen DNA besteht , oder
Zucker .
Beim Übergang von System zu System ändern sich jedoch häufig die Gleichgewichtskonstanten solcher Reaktionen, sodass wir in verschiedenen Phasen unterschiedliche tautomere Zusammensetzungen beobachten können.
Ein Beispiel hierfür ist das Histaminmolekül (siehe Abbildung unten).
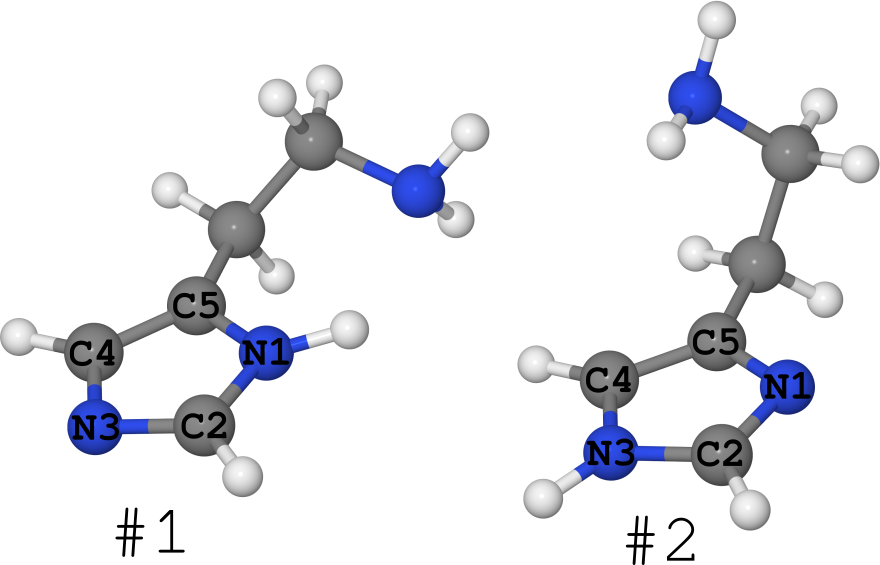
Es existiert in Form von 2 Tautomeren (ich schweige im Allgemeinen über die Anzahl der Konformere, es gibt viele von ihnen):
- # 1, wo Wasserstoff auf Stickstoff N1 sitzt,
- # 2, wo Wasserstoff auf Stickstoff N3 sitzt.
So kam es, dass für dieses Molekül seine Strukturen in verschiedenen Phasen bekannt sind.
- Im Kristall ist es in Form von # 1 vollständig "gefroren". (siehe DOI- Artikel : 10.1021 / ja00796a011 und die Struktur in der Cambridge Structures Bank unter dem Namen "HISTAN" und / oder der Nummer 1176642)
- In wässrigen Lösungen liegt dieses Molekül in beiden Formen vor und Tautomer Nr. 2 ist deutlich größer ( DOI: 10.1021 / ja027103x ).
- In Gas liegt Histamin gleichermaßen in Form Nr. 1 und in Form Nr. 2 vor ( DOI: 10.1021 / ja980560m ).
Das heißt, Unterschiedliche Phasen enthalten unterschiedliche Anzahlen unterschiedlicher Moleküle, was bedeutet, dass es sich um unterschiedliche Systeme handelt.
Fehler Schlussfolgerung # 1

Die Hauptschlussfolgerung aus den obigen Beispielen lautet wie folgt:
Wenn man Berechnungen in einer Phase mit einem Experiment in einer anderen vergleicht, muss man auf systematische Unterschiede vorbereitet sein.
Dies bedeutet nicht, dass ein Vergleich nicht erforderlich ist: Es ist ein Vergleich erforderlich, sondern es ist lediglich erforderlich, die festgestellten Unterschiede und / oder Zufälle kritischer zu betrachten und solche Auswirkungen nach Möglichkeit zu bewerten.
Fehler Nr. 2. "Zoo" molekulare Parameter.
Der zweite Fehler wird kurz wie folgt beschrieben: Wenn die Parameter ähnlich, aber nicht identisch aufgerufen werden, handelt es sich um unterschiedliche Parameter.
Um zu verstehen, woher diese Meinungsverschiedenheit zwischen Theorie und Experiment stammt, müssen sowohl die experimentellen Standardmethoden, mit denen molekulare Parameter erhalten werden, als auch die Modelle, die ähnliche Größen ausschließlich aus der Theorie berechnen, genauer analysiert werden.
Und hier werden wir wieder nur über Strukturen sprechen.
Wie man experimentelle molekulare Strukturen erhält
Um uns irgendwie einzuschränken, werden wir nur über Methoden zur Untersuchung der Struktur einzelner Moleküle sprechen, d.h. über die Gasphase.
Wir haben zwei Hauptquellen für solche Informationen:
- Gaselektronenbeugung,
- Mikrowellenspektroskopie.
Wir werden auf jede dieser Methoden näher eingehen.
Gaselektronenbeugung
Die Methode ist ziemlich alt und stammt aus den 30er Jahren des 20. Jahrhunderts, als die deutschen Wissenschaftler Mark und Wirl die ersten Experimente zur Beugung von Elektronen durch Gas durchführten.
Nur wenige wissen es, aber diese Forschungsmethode ist mit dem Erhalt von drei Nobelpreisen für Chemie verbunden.
3 Adlige mit elektronographischem Eingang- Peter Debye erhielt 1936 seine Auszeichnung mit folgendem Wortlaut:
"[für seine Arbeit an] molekularer Struktur durch seine Untersuchungen zu Dipolmomenten und der Beugung von Röntgenstrahlen und Elektronen in Gasen "
Dies ist die einzige explizite Erwähnung der Gaselektronenbeugung in den Verdiensten des Preisträgers, und das nicht ohne Grund. Die grundlegende Elektronenbeugungsgleichung für die Intensität der molekularen Streuung heißt Debye.
in der Tat die Debye-GleichungIij(s)=gij frac sin(srij)rij
Hier
Iij bezeichnet die Streuintensität von Elektronen (oder Röntgenstrahlen oder anderen Teilchen) durch ein Paar von
i- ten und
j- ten Atomen in
einiger Entfernung
rij voneinander,
s= frac2 pi lambda sin left( frac theta2 right) Ist die Streukoordinate dem Streuwinkel zugeordnet?
theta und Teilchenwellenlänge
lambda und
g - die Fähigkeit dieses Atompaares, beugende Teilchen zu streuen.
Und trotz der Tatsache, dass an diese wunderbare Physik (das Modell ionischer Lösungen , ihr Modell zur Berechnung der Wärmekapazität von Kristallen ), aber nicht an die Elektronenbeugung, alles erinnert wird, erhielt er (insbesondere) den wissenschaftlichen Hauptpreis dafür.
- Linus Pauling im Jahr 1954. Ja, derjenige, der zwei persönliche Nobelpreise erhielt
und sogar die ganze Welt mit Vitamin C versorgte, Great Pauling. Insbesondere als er in Kaltekh arbeitete, beschäftigte er sich mit Gaselektronenbeugung (siehe zum Beispiel DOI: 10.1021 / ja01873a047 ). Und natürlich half ihm das Wissen über die Strukturchemie freier Moleküle, die berühmte Theorie der chemischen Bindung zu entwickeln (aber lassen Sie uns seinen großen kristallografischen Hintergrund hier nicht herunterspielen). - Odd Hassel, Preisträger von 1969. Er erhielt seinen 1/2 Nobelpreis für die Entdeckung des Konformationsgleichgewichts. Er tat dies auf der Grundlage einer Elektronenbeugungsstudie von Cyclohexan. Dieses Molekül existiert in Form von zwei Konformationen: einem Stuhl (Stuhl) und einem Badezimmer (in der englischen Tradition - ein Boot, ein Boot).

Von hier aus: www.shapeways.com/product/N5FE298DS/cyclohexane-2-molecules-boat-and-chair-form
Diese Optionen für die Anordnung von Atomen verwandeln sich schnell ineinander, aber zu diesem Zeitpunkt wussten sie nichts davon und glaubten, dass nur eine der Strukturen realisiert werden sollte. Nur das Elektronenbeugungssignal wollte von keiner dieser Strukturen beschrieben werden, und nur eine Kombination von Signalen aus beiden Konformationen konnte das beobachtete Beugungsmuster erklären (mehr dazu in I. Khargittais Buch "Frank Science. Conversations with Famous Chemists").
Das Schema der Methode selbst ist sehr einfach (siehe Abbildung unten).
Die Sache passiert im luftleeren Raum.
- Schnelle Elektronen werden kontinuierlich aus der Kathode herausgeschlagen, die im Anodenfeld auf Energien von 40-60 keV beschleunigt werden.
- Genug gestreute (aber schnelle) Elektronen werden von einer magnetischen Linse fokussiert, wonach sie sich in einen schmalen Strahl verwandeln.
- Eine Kammer mit einer Substanz ist senkrecht zum Balken installiert. Die Probe wird zum Kochen gebracht und der entstehende Dampf kommt mit dem Elektronenstrahl in Kontakt.
- Elektronen werden erfolgreich von den Molekülen gestreut und fliegen leise weiter weg, wo sie auf den Film fallen.
- In der Regel vor den Film stellen die sogenannten. Sektor Gerät. Dies ist ein sehr schnell rotierender Bildschirm mit ungewöhnlicher Form. Tatsache ist, dass das Elektron die Wahrscheinlichkeit hat, von seiner ursprünglichen Richtung abzuweichen (um einen großen Streuwinkel t h e t a ), fällt sehr schnell. Um diese Abnahme der Intensität auszugleichen, überschattet der Sektor daher gleichmäßig den zentralen Teil des Films und lässt den fernen Teil offen. Das Ergebnis ist ein gleichmäßigeres Bild.
- Die Strahlfalle fängt die Elektronen auf, die überhaupt nicht gestreut sind (und es gibt viele davon).
- Nun, damit die Moleküle nicht über das gesamte Gerät fliegen und es verschmutzen, werden sie in einer mit flüssigem Stickstoff gekühlten Kühlfalle eingefroren.
Das Ergebnis ist das gleiche Beugungsmuster konzentrischer Ringe, das durch die Debye-Gleichung beschrieben wird (dies ist ein Signal). Verschiedene molekulare Parameter können dann direkt daraus gezogen werden.
Wo finde ich Gaselektronenlabore?Es sind nicht mehr so viele übrig.
In Russland gibt es jedoch zwei davon: Moskau (am Chemischen Institut der Moskauer Staatlichen Universität) und an der Chemisch-Technologischen Universität von Iwanowo.
Mikrowellenspektroskopie
Diese Methode zur Untersuchung von Molekülen ist bekannter, daher werde ich kurz darauf eingehen und die modernste Modifikation als Beispiel verwenden: ein Fourier-Transformationsspektrometer (wie auf Russisch, kurz Fourier-transformierte Mikrowellenspektroskopie).
Das Design hier ist bereits komplizierter, da es eine Reihe verschiedener Elektronik (Verstärker, Frequenzmodulatoren usw.) erfordert. Wir werden das alles weglassen und nur darüber sprechen, was in der Vakuumkammer passiert.
- Gegenüber stehen zwei Hornantennen (wie die, mit der die Reliktstudie eröffnet wurde). Einer von ihnen dient als Sender und der zweite als Empfänger.
- Senkrecht zu diesen Antennen befindet sich ein Ventil, das die Probe startet. Meistens wird es in Form von Dampf zusammen mit einem bestimmten Trägergas (normalerweise Inertgase) im adiabatischen Expansionsmodus gestartet. Unter solchen Bedingungen kühlen die Moleküle schnell auf Temperaturen nahe 0 K ab, was das Spektrum stark vereinfacht und es anfälliger für Interpretationen macht.
- Wenn Moleküle die gesamte Kammer füllen, bestrahlt die Sendeantenne sie mit einem linearen frequenzmodulierten Signal. In der Frequenzdarstellung entspricht dies der Summe aller Frequenzen in einem bestimmten Bereich.
- Einige Moleküle absorbieren diese Strahlung bei verschiedenen Sendefrequenzen und gehen in einen angeregten Zustand über. Aber nach einiger Zeit fallen sie zurück und beginnen zu strahlen, was sie während des Impulses von der Sendeantenne gefangen haben. Dieser Stillstand sieht aus wie ein abnehmendes Schwingungssignal ( freier Induktionsabfall ). Die zweite Antenne registriert es ebenfalls. Dann wird nach der Fourier-Transformation der zeitlichen Aufzeichnung dieses Signals das übliche Frequenzspektrum erhalten.
Im Gegensatz zur Elektronenbeugung, die bei der Mikrowellenspektroskopie nicht wichtig war, welche Art von Molekülen zu berücksichtigen ist, muss ein Molekül ein konstantes Dipolmoment aufweisen (in seltenen Fällen ist auch ein magnetisches Dipolmoment geeignet, dies ist typisch für Radikale wie ein O
2 -Molekül). Das Signal hier ist „Emissionsintensität vs. Frequenz. " Aus diesen Spektren werden Rotationskonstanten durch einige Modelle extrahiert, aus denen dann die Molekülstruktur extrahiert wird.
Willkommen im Zoo der molekularen Parameter!
Jetzt ist es Zeit zu untersuchen, welche geometrischen Parameter wir aus verschiedenen Experimenten erhalten können. Tatsächlich gibt jede der Arten von Größen an, welche Art von Modell verwendet wurde, um das experimentelle Signal anzupassen (meistens nach der Methode der kleinsten Quadrate). Die meisten dieser Parameter finden sich in der Übersicht Kuchitsu K., Cyvin SJ // In: Molekulare Struktur und Schwingungen / Cyvin SJ (Hrsg.) - Amsterdam: Elsevier, 1972.- Ch.12. - S.183-211.
Beginnen wir noch einmal mit der Elektronographie.
- rg= langler rangleT Struktur. Dies ist nur eine Reihe von Durchschnittswerten der interatomaren Abstände bei einer bestimmten Temperatur.
- ra= langler−1 rangle−1T Dieser Wert ist ähnlich rg , aber es ist etwas natürlicher für die Beschreibung des Beugungsmusters.
- r alpha=rh,0=? . Dieser Wert hat keine klare physikalische Bedeutung und ist vollständig an das Interpretationsmodell gebunden. Tatsächlich wird dies in der Kristallographie beobachtet.
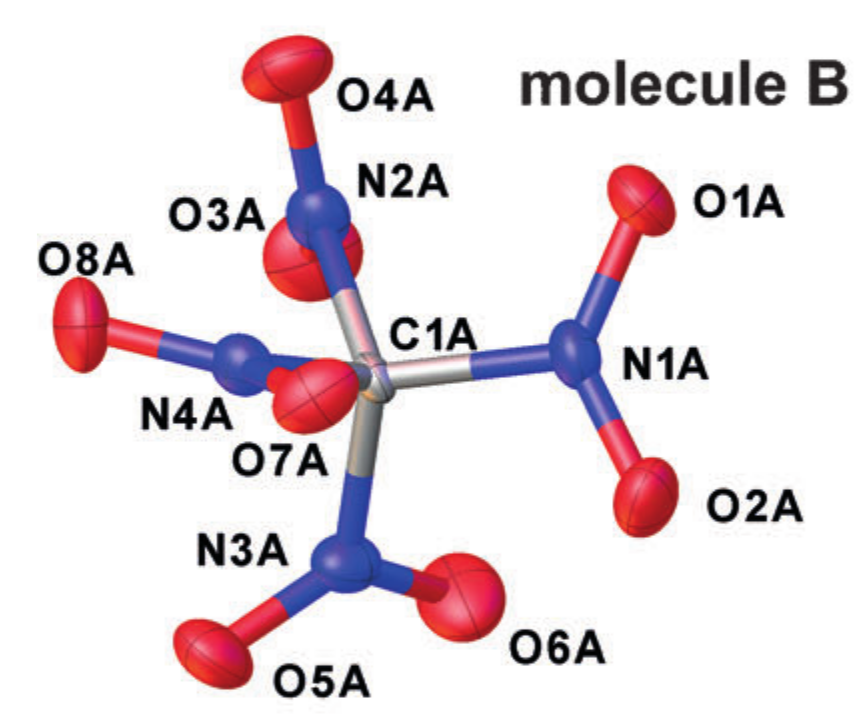
Kristallographisches Beispiel r alpha Strukturen (Tetranitromethan im Kristall). Angepasst von DOI: 10.1002 / anie.201704396
Jedes Atom wird durch ein Ellipsoid angenähert, das seine Schwingungsbewegung beschreibt, und die Abstände zwischen den Zentren der resultierenden Ellipsen werden als interatomare Abstände genommen. Eine solche Vereinfachung der Art der Bewegung von Atomen entspricht jedoch der Einführung der harmonischen Oszillator-Näherung für Schwingungen und funktioniert nicht immer gut.
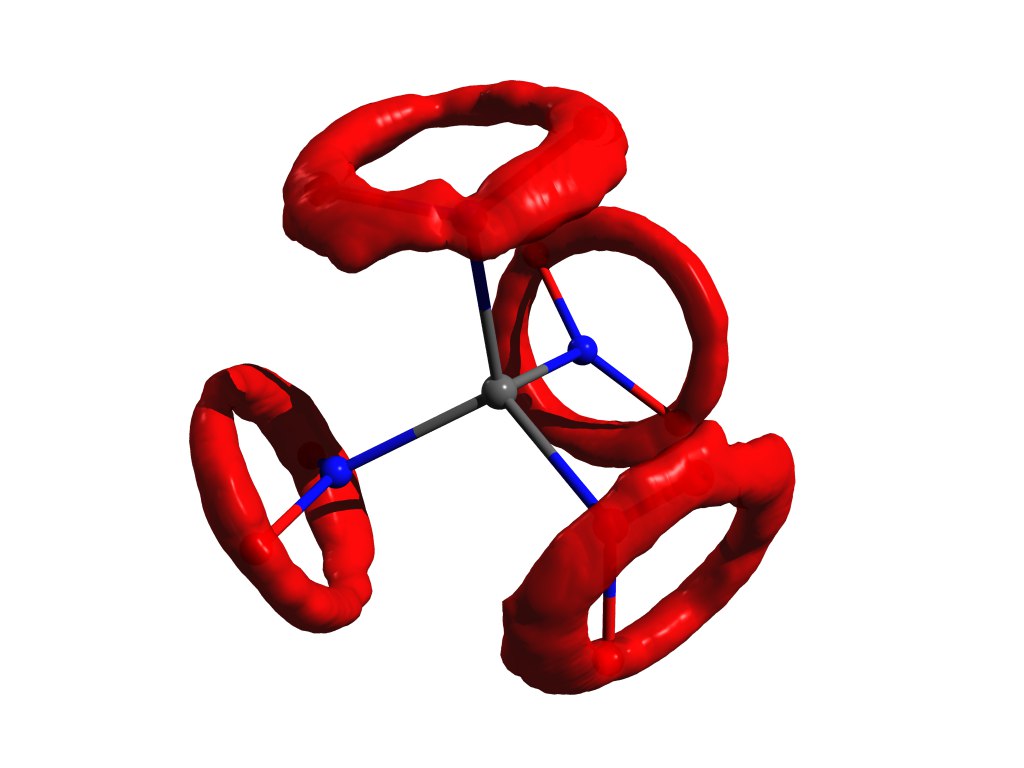
Ein Beispiel für die Verteilung von Atomen, wenn die Näherung des harmonischen Oszillators nicht funktioniert. Das gleiche Tetranitromethan, aber im Gas.
- rh1=??? . Dieser
HEX- Wunder-Yudo-Fischwal wird nicht auf den Punkt gebracht, er hat eine sehr schwache Beziehung zur Realität, aber er hat wunderbare Eigenschaften: Er muss geometrisch konsistent sein (siehe unten) und kann leicht berechnet werden. Aufgrund dessen gewann sie ihre große Popularität in der elektronografischen Gemeinschaft.
In der Mikrowellenspektroskopie gibt es etwas weniger Strukturvarianten.
- Das physikalisch verständlichste ist rn= langlen| hatr|n rangle Struktur. Tatsächlich ist dies die gemittelte Geometrie über einen bestimmten Schwingungszustand des Moleküls ( |n rangle ) Da sie am häufigsten mit kalten Molekülen arbeiten, beobachten sie normalerweise eine bestimmte Struktur aus dieser Klasse: r0 d.h. die Geometrie des Moleküls im Grundschwingungszustand, wenn Atome um ihre vorteilhafteste Position keine Schwingungen erzeugen.
- Am beliebtesten ist rs Struktur. Der Index "s" bedeutet "Substitution". Sie verstehen es so: Es wird angenommen, dass es einige Koordinaten von Atomen gibt, die im Raum festgelegt sind, dann machen sie eine Einzelatom-Isotopensubstitution eines Atoms im Molekül und bestimmen die Position dieses Atoms durch Ändern der Rotationskonstanten. Der Hauptvorteil dieser Technologie ist die Einfachheit. Minus: Sie brauchen nur Monosubstitution + nicht alle Positionen von Atomen können so festgelegt werden + die physikalische Bedeutung eines solchen Modells ist nicht sehr klar.
- Logische Entwicklung rs Strukturen sind rm -Strukturen, die durch Anpassen durch massengewichtete kleinste Quadrate erhalten werden. Sie benötigen auch isotopensubstituierte Moleküle, aber jedes von ihnen ist bereits geeignet.
Und das ist weit entfernt von allen möglichen Arten von Strukturen ...
Aber der
große Simulator Der Benutzer eines quantenchemischen Standardpakets (wie des
Gaussian Evil Corporation-Programms ) erhält bei Verwendung eines Zaubers wie „Opt“ die sogenannte „Gleichgewichtsgeometrie“ oder
re Struktur. Dies ist die optimalste Konfiguration der Kerne, wodurch die elektronische Energie des Systems minimiert wird. Und solche Strukturen können auch aus der Elektronenbeugung und Rotationsspektroskopie herausgezogen werden, jedoch nur für sehr kleine und symmetrische Moleküle und in Kombination mit anderen Forschungsmethoden. Bisher klappt es nicht.
Und so stellt sich die Frage: Ist es richtig zu vergleichen?
re Struktur mit einigen der experimentellen, nur die experimentellen Fehler betrachten?
Die Antwort hier ist einfach:
Nein , es ist notwendig, einen zusätzlichen Fehler auf mögliche systematische Unterschiede zu legen. Ein sehr anschauliches Beispiel hierfür ist der Bastiansen-Morino-Effekt (siehe Artikel
DOI: 10.1107 / S0365110060002557 und
DOI: 10.1107 / S0365110060002545 ).
Angenommen, wir haben ein Molekül vom Typ CX
2 (d. H. CO
2 , CS
2 usw.). Wie wir aus der Schulchemie wissen sollten, haben diese Moleküle eine lineare Struktur (Kohlenstoffatome und zwei X-Chalkogene liegen auf einer geraden Linie).
Dies bedeutet, dass der Abstand zwischen den X-Atomen gleich der doppelten Länge der C-X-Bindung sein sollte (d. H.
re( mathrmXX)=2re( mathrmCX) )

Wie auch immer, wenn wir die Abstände zwischen den Atomen C und X messen (
rg( mathrmCX) ) und XX (
rg( mathrmXX) ) durch Gaselektronenbeugung erhalten wir das
rg( mathrmXX)<2rg( mathrmCX) d.h. Das Molekül ist gekrümmt. Der Grund liegt in der Tatsache, dass das Molekül das sogenannte macht
Scherenschwingungen , aufgrund derer die X-Atome viel näher beieinander liegen als an der günstigsten Stelle (siehe Abbildung unten).

Woher kommt der Bastansen-Morino-Effekt? Bild aus dem
DOI- Artikel
: 10.1039 / C6CP05849C .
Wenn wir also den Temperaturmittelwert gleichsetzen
rg -Struktur zum Gleichgewicht (
re ) würden wir die falsche Schlussfolgerung ziehen, dass die Moleküle Kohlendioxid und Schwefelkohlenstoff gekrümmt sind.
Deshalb müssen Sie beim Vergleich verschiedener Arten von geometrischen Parametern immer sehr vorsichtig sein. Dies gilt sowohl für einen Vergleich experimenteller Daten untereinander als auch für einen Vergleich von Experiment und Theorie.
Moleküle Standardmodellmoleküle
Stellen wir uns nun vor, wir wollten von ganzem Herzen das Ergebnis eines Experiments auf der Grundlage unseres theoretischen Modells simulieren, um die Simulation mit der Realität im fairen Kampf zu vergleichen.
Und hier ist es auch notwendig, vorsichtig zu sein, weil Verschiedene Modelle von Molekülen haben auch ihre Grenzen der Anwendbarkeit. Lassen Sie uns dies am Beispiel des Standardmolekülmodells untersuchen.
Zuerst müssen Sie verstehen, was das Standardmodell des Moleküls ist. BAK-Physiker haben ein eigenes
Standardmodell , Astronomen
ein eigenes und Physiker ein eigenes Grunddesign, nach dem sie später tanzen. Im Gegensatz zu physischen Modellen betrachten wir jedoch eine Reihe von Näherungswerten, mit denen der Benutzer das Ergebnis relativ automatisch und schnell erhalten kann.
Für Gaußsche BenutzerNun erinnern wir uns, was den Gaußschen Zaubersprüchen „Opt“ und „Freq“ zugrunde liegt.
Das allgemeine Schema der eingeführten Näherungen sieht ungefähr so aus:
Ganz unten in der Qualität steht unser Standardmodell. Gehen Sie kurz alle Phasen des Eingangs durch.
Das resultierende Modell heißt RR-HO (@BO). Wir werden die Born-Oppenheimer (BO) -Näherung nicht berühren, aber wir müssen im Rahmen der Strukturchemie über den harten Rotator und den harmonischen Oszillator sprechen ...Und das Hauptproblem bei dieser Näherung ist, dass das Molekül nicht starr ist und seine Schwingungen vollständig harmonisch sind. Dementsprechend benötigen wir in der Realität die Annäherung eines nicht festen Rotators und eines anharmonischen Oszillators. Und das Schlüsselwort hier ist "anharmonisch", d.h. "Nicht harmonisch."Sprechen wir über die einfachsten Moleküle: zweiatomig. Es gibt viele Beispiele dafür: HCl, HBr, HI, CO, O 2 , N 2 usw. usw.
Sie unterscheiden sich von allen Molekülen dadurch, dass sie nur eine Schwingung haben: die Ausdehnung / Kompression des interatomaren Abstandes.Und dies ist der Abstand zwischen den Atomen, den wir bei der Gaselektronenbeugung messen können (in einer Variante der Durchschnittstemperatur,r g ) und in der Rotationsspektroskopie (gemittelt über beispielsweise den Grundschwingungszustand, d.h.r 0 )
Und jetzt stellt sich die Hauptfrage des Universums des Lebens und allgemein:was wird sein r g und r 0 in der Näherung eines harmonischen Oszillators und wie korreliert dies mit dem Gleichgewichtsabstandr e ?
Um eine Antwort zu erhalten, müssen Sie die Oberfläche der potentiellen Energie für ein zweiatomiges Molekül betrachten:
- , : . , .
- , , 2 :
- ( r→0 ), - , « » ,
- ( r→+∞ )
Wenn das Molekül seine Bindungslänge relativ zur Gleichgewichtsposition verkürzt, liegt es an der Wand an und fällt auf ein weiches Sofa, wenn es zunimmt. Und das Molekül ist kein Dummkopf, es wird mehr auf der Couch liegen als gegen die Wand schlagen. Daher ist die über die Entfernung gemittelte Schwingung größer als das Gleichgewicht (r e < r 0 , r g ), und dies fällt auf: Solche Verschiebungen liegen in der Größenordnung von 0,01 Å, was höher ist als die Messfehler.
Selbst wenn wir so etwas wie ein Experiment berechnen möchten, das im Rahmen des Standardmolekülmodells (RR-HO @ BO) verbleibt, erhalten wir daher nichts Neues. Daher wird die Gleichgewichtsgeometrie am Vergleich teilnehmen.Fehler Schlussfolgerung # 2
 Abbildung aus dem DOI- Artikel : 10.1002 / anie.201611308 .Und die Schlussfolgerung ist furchtbar einfach und besteht aus 2 Teilen.
Abbildung aus dem DOI- Artikel : 10.1002 / anie.201611308 .Und die Schlussfolgerung ist furchtbar einfach und besteht aus 2 Teilen.- Bei einem korrekten Vergleich sollten alle Werte die gleiche Bedeutung haben.
- Wenn die Werte unterschiedlich sind, sollte dies nicht vergessen werden.
Beispiele für Fehler in wissenschaftlichen Arbeiten
"Hindu arbeitet"
Tatsächlich ist der Hauptort, an dem Sie dies finden können, Low-Level-Magazine. Sie enthalten selten Artikel mit coolen Ergebnissen, daher wurden sie von führenden Forschern aus Ländern der zweiten und weiteren Welten (BRICS-Länder und ihre weniger erfolgreichen Anhänger) ausgewählt. Mit "Low-Level" -Magazinen sind hier nicht diejenigen gemeint, die Artikel wie "The Rooter: Algorithmus für die typische Vereinheitlichung von Zugangspunkten und Redundanz" veröffentlichen , sondern angesehene wissenschaftliche Veröffentlichungen. In meinem wissenschaftlichen Bereich sind die bekanntesten „Halbwäschen“:(Es gibt andere). Wie Sie sehen können, gelten formale Zeichen in der russischen Wissenschaft als sehr anständige Veröffentlichungen. Aber es kommt ein Ansturm von g ... Inhalt von fragwürdiger Qualität, dass noch viel undicht.Zur Veranschaulichung habe ich die neueste Ausgabe des Journal of Molecular Structure genommen und das Inhaltsverzeichnis durchgesehen, und voila:S. Sathiya, M. Senthilkumar, C. Ramachandra Raja, Kristallwachstum, Hirshfeld-Oberflächenanalyse, DFT-Studie und NLO-Studien dritter Ordnung von Thioharnstoff-4-dimethylaminobenzaldehyd // J. Mol. Gen. Struct., V. 1180 (2019), PP. 81-88.
https://doi.org/10.1016/j.molstruc.2018.11.067
Die allgemeine Struktur solcher Arbeiten ist sehr unprätentiös.- Einige Substanzen werden „gekocht“ (aber häufiger dumm bei Sigma gekauft ). In dieser Arbeit wird die Substanz noch gekocht.
- (), . — , ( , ) , . , - Gaussian, « ». … 1 2 , .. - .
- , /Vis, .
- In molekularen Standardvisualisierern wie (grunzenden) GaussView werden schöne Bilder gezeichnet, jedoch immer in schlechter Qualität.
- Es werden keine wesentlichen Schlussfolgerungen gezogen: „Wir haben viel experimentiert, viel gezählt, Tabellen und Bilder mitgebracht ⇒ Wir sind großartig, gib uns Süßigkeiten. “
Aber von meinen Favoriten: ArtikelM. Govindarajan, M. Karabacak, FT-IR-, FT-Raman- und UV-Spektraluntersuchung; Berechnete Frequenzschätzungsanalyse und elektronische Strukturberechnungen an 1-Nitronaphthalin // Spectrochimica Acta Teil A: Molekulare und biomolekulare Spektroskopie, V. 85 (2012), PP. 251-260,
https://doi.org/10.1016/j.saa.2011.10.002.
, HO . , ,
.
, « » ( "
") , .
,
, . «-» (c/ «»)
Kommt es in guten Zeitschriften vor?
Ja, da gibt es „Fehler“.Hab keine Angst, alles endete gut!, , , / . , , .
, : , .
Wir sprechen von einem der organischen Moleküle mit einer extrem langen CC- Einfachbindung: 1,1'-Bisdiadamantan :
Warum ist dieses Molekül cool?,
(
), , C--C 1.54 Å.
, 1,1'-
re=1.630±0.005 Å, 0.08 Å ( )!
C--C - , , , , . , , () . - , . , ( ) , .
Die zentrale unäre Verbindung CC ist sehr lang und daher war es sehr interessant zu vergleichen, wie theoretische Methoden die Realität reproduzieren. Und die Realität ist in den ersten Artikeln, hier sind sie:
Holy shit!— Nature, , — JACS — !!! , - .
wurde nur durch kristallographische Daten dargestellt. Als Ergebnis des Vergleichs schwieriger vergleichbarer Dinge miteinander, ohne die Unterschiede in den Parametern richtig zu korrigieren, kamen sie schließlich zu dem Schluss, dass die Länge der zentralen CC-Bindung im Gas 1,655 Å beträgt und um 0,02 Å überschritten wird. Und das ist deutlich mehr als der experimentelle Fehler.Glücklicherweise haben sie am Ende mit Spezialisten in diesen Fragen zusammengearbeitet und am Ende die richtige Antwort erhalten (eine kurze populäre Zusammenfassung dieser Arbeit finden Sie auch auf N + 1 ).Benötigen Sie einen Vergleich?
 Nach allem, was ich über die Richtigkeit der Vergleiche geschrieben habe, kann sich eine vernünftige Frage stellen: Ist es dann notwendig, die Ergebnisse von Berechnungen und die Ergebnisse von Experimenten untereinander zu vergleichen?Es ist notwendig! Genau wie Sie brauchen!Es gibt eine berühmte Aussage (von der ich keinen zuverlässigen Autor finden konnte):
Nach allem, was ich über die Richtigkeit der Vergleiche geschrieben habe, kann sich eine vernünftige Frage stellen: Ist es dann notwendig, die Ergebnisse von Berechnungen und die Ergebnisse von Experimenten untereinander zu vergleichen?Es ist notwendig! Genau wie Sie brauchen!Es gibt eine berühmte Aussage (von der ich keinen zuverlässigen Autor finden konnte):Niemand glaubt theoretischen Berechnungen, außer dem, der sie gemacht hat.
Jeder glaubt an experimentelle Ergebnisse, außer dem, der sie erhalten hat.
Übersetzt ins Russische klingt es so: Niemand glaubt den theoretischen Berechnungen, außer dem, der sie gemacht hat, aber jeder glaubt den experimentellen Daten, außer dem, der sie erhalten hat.Aber in der Wissenschaft ist es notwendig, dass jeder glaubt (naja oder die Mehrheit), und Experiment ist das einzige Maß, da es das, was wir berechnet haben, mit der Realität in Beziehung setzt.In der zweitwichtigsten chemischen Zeitschrift gibt es einen wunderbaren Aufsatz (Open Access) zum Thema, was ein Forscher oder eine durchschnittliche Person, die wissenschaftliche Artikel und / oder Nachrichten liest, tun sollte: Mata R., Suhm M. // Angew. Chem. Int. Ed., 56 (2017), DOI: 10.1002 / anie.201611308(ich habe übrigens bereits einen Link dazu gegeben, da ein Bild von Ricardo Mata aus diesem Artikel stammt).Die Schlussfolgerungen dieses Aufsatzes geben Empfehlungen an Simulationstheoretiker und Experimentatoren. Ich werde sie hier (in Übersetzung und einer kleinen Überarbeitung) als letztes Wort zu diesem Beitrag geben.- Der Theoretiker muss:
- Geben Sie nicht nur erfolgreiche Methoden und Versuche an, sondern beschreiben Sie auch die Fehler von Methoden (insbesondere wenn diese Methoden beliebt sind).
- Beschreiben Sie Ihre Methodik gut und vollständig.
- wo es möglich ist, Schätzungen (oder Beschreibungen) von Fehlern und wichtige akzeptierte Annäherungen und Vereinfachungen anzugeben.
- Die Experimentatoren müssen wiederum:
- Theoretiker in ihre experimentellen Daten zu stecken, die sie als Benchmark (Standards) verwenden könnten,
- um der wissenschaftlichen Gemeinschaft unverständliche experimentelle Daten zu zeigen, bei denen die Theorie (oder zusätzliche Experimente) bei der Erklärung helfen würde,
- auf
fremdem Gebiet von theoretischen Konferenzen mit ihren Daten zu sprechen ,
persönliche Geschichte, , , .
- Ziehen Sie aus Ihren experimentellen Daten Dinge heraus, die für den Vergleich mit der Theorie so zugänglich wie möglich sind.
Alles Gute und korrekte Vergleiche! Und denken Sie daran: Nur wer nichts tut, irrt sich nicht.
PS
Als Nachwort möchte ich eine kleine Liste von Datenbanken geben, in denen Sie verschiedene experimentelle Daten für Moleküle ausgraben können.Strukturelle Datenbanken
Kristallographische Dosen
Der einfachste Weg, die Struktur eines Moleküls in einem Kristall zu bestimmen, ist weil PCA ist eine Routine. Wenn Sie nicht wissen, wie das Molekül aussieht, gehen Sie zu den kristallografischen Datenbanken (Orte, an denen fast alle Strukturen von Substanzen gesammelt werden, die jemals in ein Goniometer gepfercht und von einem Strahl kurzwelliger Partikel beleuchtet wurden). Da es viele solcher Banken gibt, werde ich nur die bekanntesten nennen (eine vollständigere Liste finden Sie im Wiki ).- Anorganische Kristallstrukturdatenbank ( ICSD ). Hier geht es nicht nur um Moleküle, dort findet man hauptsächlich Strukturen verschiedener Salze, Metalle, Keramiken usw. Diese Basis wird von der Technischen Universität Karlsruhev unterstützt, daher ist der Zugang dazu kostenpflichtig und nicht billig. Aber wenn das so ist, ihre Seite .
- Cambridge Structural Database ( CSD ). , . ! . ! Zum Hinzufügen. , , , , . .
- Crystallography Open Database ( COD ). - . , , , , . .
- , , Protein Data Bank ( PDB ). , ( ). .
Die Struktur von Molekülen in einem Gas
Hier ist es etwas schlimmer, weil Experimente zur Untersuchung der Struktur freier Moleküle sind viel komplizierter, sowohl zur Durchführung (mindestens ein Hochvakuum ist erforderlich) als auch zur Interpretation.Daher gibt es wesentlich weniger Datenbanken.- Die größte Datenbank für freie Moleküle ist die MOlecular GAsphase DOCumentation (oder MOGADOC ). Es hat seinen Sitz an der Universität Ulm und ist eine sehr teure Investition. Aber wenn überhaupt, ist die Seite hier .
- Wenn Sie die 100% experimentellen Gleichgewichtsstrukturen von Molekülen kennen möchten, dann ist dies die NIST Computational Chemistry Comparison and Benchmark DataBase ( CCCBDB ). Fast alle rein experimentellre - , . .
- — MolWiki . , , , ( ). .
?
 Entnommen aus xkcd.comHier gibt es deutlich mehr Datenbanken, da das Entfernen des Spektrums ohne Interpretation eine viel einfachere Aufgabe ist als das Erhalten einer Struktur (Sie müssen keine Modelle erstellen und beweisen, dass sie korrekt sind). Außerdem sind die Spektren von großem Wert: Sie können verwendet werden, um die Zusammensetzung von Proben zu bestimmen, sei es Wasser aus einem benachbarten Fluss oder ein Signal aus einer Molekülwolke (oder sogar aus einer Exoplanetenatmosphäre, siehe Abbildung oben).Ja, alle Links in diesem Abschnitt führen zu kostenlosen Datenbanken.
Entnommen aus xkcd.comHier gibt es deutlich mehr Datenbanken, da das Entfernen des Spektrums ohne Interpretation eine viel einfachere Aufgabe ist als das Erhalten einer Struktur (Sie müssen keine Modelle erstellen und beweisen, dass sie korrekt sind). Außerdem sind die Spektren von großem Wert: Sie können verwendet werden, um die Zusammensetzung von Proben zu bestimmen, sei es Wasser aus einem benachbarten Fluss oder ein Signal aus einer Molekülwolke (oder sogar aus einer Exoplanetenatmosphäre, siehe Abbildung oben).Ja, alle Links in diesem Abschnitt führen zu kostenlosen Datenbanken.- NIST Chemistry WebBook . , . , UV/Vis, - . , ! , , . .
- High-resolution transmission molecular absorption database ( HITRAN ). - , , , , (, ). .
- , The Cologne Database for Molecular Spectroscopy, . .
- Nun, als letztes Beispiel werde ich das ExoMol- Projekt geben . Dies sind keine rein reinen experimentellen Spektren, aber dies ist ein hervorragendes Beispiel für die Wechselwirkung von Theorie und Experiment: Basierend auf hochpräzisen experimentellen Daten und Berechnungen auf sehr hohem Niveau werden die Spektren einfacher Moleküle unter verschiedenen (einschließlich extremen) Bedingungen vorhergesagt. Das Hauptaugenmerk liegt hier auf Biomarkern. Wenn Astronomen die Spektren von Exoplaneten sehen, können sie die Moleküle, die wir bereits in ihnen kennen, leicht identifizieren. Die Seite .
PPS
Wenn es Fehler gibt / etwas unverständlich bleibt, schreibe in die Kommentare - ich werde korrigieren / versuchen, es besser zu erklären.