Vor nicht allzu langer Zeit habe ich den Science-Fiction-Roman
„Das Drei-Körper-Problem“ von Liu Qixin gelesen. Darin hatten einige Außerirdische ein Problem - sie wussten nicht, wie sie mit ausreichender Genauigkeit die Flugbahn ihres Heimatplaneten berechnen konnten. Im Gegensatz zu uns lebten sie in einem Drei-Sterne-System, und das „Wetter“ auf dem Planeten hing stark von ihrem relativen Standort ab - von der Verbrennungshitze bis zur eisigen Kälte. Und ich habe mich entschlossen zu prüfen, ob wir solche Probleme lösen können.
Physik des Phänomens
Um das Problem zu verstehen, muss man sich mit der Physik des Phänomens befassen. Im Rahmen der klassischen Theorie wird die Anziehungskraft zweier Körper durch das
Newtonsche Gesetz bestimmt:
v e c F ( V e c r 1 , v e c r 2 ) = - G m 1 m 2 f r a c v e c r 1 - v e c r 2 | v e c r 1 - v e c r 2 | 3 ,
wo
vecr1, vecr2 - die Position von Körpern im Raum,
m1,m2 - Massen von Körpern,
G - Gravitationskonstante.
Im System von
N Körper auf jedem von ihnen werden durch die Anziehungskraft des Restes beeinflusst, die durch die Gleichung ausgedrückt wird:
vecFn=−G sumk neqnmnmk frac vecrn− vecrk| vecrn− vecrk|3.
Mit
dem zweiten Newtonschen Gesetz schreiben wir die Beschleunigung für jedes Teilchen:
vecan= vecFn/mn=−G sumk neqnmk frac vecrn− vecrk| vecrn− vecrk|3.
Wenn wir uns daran erinnern, dass die Beschleunigung die zweite Ableitung der Koordinate ist, erhalten wir eine partielle Differentialgleichung zweiter Ordnung, die gelöst werden muss, um die Flugbahn jedes Körpers zu erhalten:
frac partiell2 vecrn partiellt2=fn=−G sumk neqnmk frac vecrn− vecrk| vecrn− vecrk|3.
Hierbei ist zu beachten, dass die Komplexität der Berechnung einer Funktion
fn ist gleich
O(N2) und nimmt mit zunehmender Anzahl interagierender Körper signifikant zu.
Mathe
Die erste und einfachste Methode zum Lösen von Differentialgleichungen ist
die Euler-Methode , mit der Gleichungen der folgenden Form gelöst werden können:
fracdydx=f(x,y).
Beim Übergang in die diskrete Region erhalten wir:
yi=yi−1+hf(xi−1,yi−1), quadi=1,2,3, dots,m,
wo
h Ist der Integrationsschritt und
m - die Anzahl der Integrationsschritte. Also, wenn wir die Position der Körper gleichzeitig berechnen müssen
T dann sollten wir tun
m=T/h Integrationsschritte. Hier ist das erste Problem sichtbar - wenn
T groß, dann müssen wir eine große Anzahl von Integrationsschritten machen.
Um die Euler-Methode auf unser Problem anzuwenden, sollte sie auf ein System erster Ordnung reduziert werden. Dazu führen wir eine zusätzliche Variable ein - die Partikelgeschwindigkeit:
frac partiell vecvn partiellt=fn, frac partiell vecrn partiellt= vecvn.
Das zweite Problem bei der Lösung von Differentialgleichungssystemen ist die Genauigkeit der Lösung und ihrer Steuerung. Die Genauigkeit kann auf zwei Arten verbessert werden: durch Verringern des Integrationsschritts und Auswählen einer Methode mit einer höheren Genauigkeitsordnung. Beide Methoden führen zu einer erhöhten Rechenkomplexität, jedoch auf unterschiedliche Weise. Sie können beispielsweise die klassische
Runge-Kutta-Methode vierter Ordnung verwenden , für die vier Funktionsberechnungen erforderlich sind
fn bei jedem Schritt, hat aber eine Reihenfolge der Genauigkeit
O(h4) (Zum Vergleich hat die Euler-Methode eine Reihenfolge der Genauigkeit
O(h) und erfordert eine Berechnung
fn ) Die Genauigkeit der Lösung kann auch auf verschiedene Arten gesteuert werden: Vergleichen Sie mit der analytischen Lösung, lösen Sie mit verschiedenen Methoden oder mit verschiedenen Schritten und vergleichen Sie die Ergebnisse, kontrollieren Sie Parameter und Einschränkungen von Drittanbietern, denen die Lösung entsprechen muss.
Jedes dieser Verfahren hat auch seine Nachteile. Analytische Entscheidungen können fehlen oder sogar in den meisten Fällen vollständig fehlen. Zum Beispiel für unsere Aufgabe
N tel analytische Lösung ist nur für
N=2 Aber auch das reicht aus, um die Genauigkeit der Methoden zu testen. Das Lösen eines Problems mit zwei Methoden oder mit unterschiedlichen Schritten erhöht die Rechenzeit, aber dieser Ansatz kann auf fast jede Aufgabe angewendet werden. Nicht jede Aufgabe hat Einschränkungen, aber für unsere haben sie sie: Bei jedem Integrationsschritt können wir die Umsetzung
von Naturschutzgesetzen kontrollieren. Dieser Ansatz erhöht auch die Komplexität der Berechnung, es gibt jedoch eine große Auswahl: Die Berechnung der Summe der Impulse oder Drehimpulse aller Partikel hat eine Komplexität in der Größenordnung
O(N) , während die Berechnung der Gesamtenergie eines Systems Auftragskomplexität hat
O(N2)Hinweis zur Berechnung der GesamtenergieIn unserem Fall besteht die Gesamtenergie des Systems aus zwei Teilen - kinetischer und potentieller Energie. Kinetische Energie besteht aus der Summe der kinetischen Energien aller Körper. Um die potentielle Energie zu berechnen, müssen wir die potentiellen Energien jedes Teilchens im Gravitationsfeld der verbleibenden Teilchen addieren, also müssen wir addieren
O(N2) Begriffe. Die Schwierigkeit besteht darin, dass alle Terme eine sehr unterschiedliche Reihenfolge haben und es selbst bei Berechnungen mit doppelter Genauigkeit nicht möglich ist, diesen Wert mit einer Genauigkeit zu berechnen, die für den Vergleich in verschiedenen Schritten ausreicht. Um dieses Problem zu lösen, war es notwendig, eine Summation gemäß
dem Kahan-Algorithmus anzuwenden.
Abb. 1: Ein Beispiel für eine elliptische Flugbahn.
Betrachten Sie den einfachen Fall eines Satelliten, der sich in einer elliptischen Umlaufbahn um die Erde bewegt. Wenn sich der Satellit der Erde nähert, nimmt seine Geschwindigkeit zu, und wenn er sich von der Erde entfernt, nimmt er ab. Dementsprechend besteht die Möglichkeit, den zeitlichen Integrationsschritt im grünen Teil der Umlaufbahn zu verringern und ihn in Rot und Blau zu erhöhen, ohne die Genauigkeit der Lösung zu ändern. Versuchen wir es genauer zu vergleichen.
Tabelle 1. Untersuchte Methoden zur Lösung von DifferentialgleichungenUm die beste Methode für unsere Aufgabe auszuwählen, werden wir einige bekannte Methoden vergleichen. Dazu simulieren wir die Kollision zweier Körpersysteme
N=512 und messen Sie die relative Änderung der
Gesamtenergie , des
Impulses und ihres
Moments am Ende der Simulation (maximale Simulationszeit)
Tmax=2,5 ) In diesem Fall variieren wir den Schritt und die Parameter der Integrationsmethoden und messen die Anzahl der Funktionsaufrufe
fn Diese Methoden, die bei einer geringeren Anzahl von Anrufen zu weniger Verlusten führen, werden wir als akzeptabler betrachten.

| 
|
| a) | b) |
| Abbildung 2. Relative Änderung der Energie a), Impuls b) am Ende der Systemsimulation N=512 Körper nach verschiedenen Methoden in Abhängigkeit von der Anzahl der Funktionsberechnungen fn doppelte Präzision |
Aus den Diagrammen von 2 ist ersichtlich, dass das beste Verhältnis der Menge der Funktionsberechnung
fn und die relative Änderung der Energie des Körpersystems in den Adams- und Dorman-Prince-Methoden fünfter Ordnung. Es ist auch ersichtlich, dass für alle Methoden mit einer Zunahme der Anzahl von Berechnungen
fn Die relative Änderung des Impulses des Systems nimmt zu. Bei einer relativen Energieänderung macht sich dies ebenfalls bemerkbar, jedoch nur bei wenigen Methoden, die die Schwelle erreichen könnten
dE/E0<10−12 . Dieser Effekt kann nicht mehr auf den Fehler von Methoden zurückgeführt werden, sondern auf den Fehler von Berechnungen, und eine weitere Erhöhung der Genauigkeit ist nur zusammen mit einer Erhöhung der Genauigkeit von Berechnungen mit Gleitkomma möglich.

| 
|
| a) | b) |
| Abb. 3. Relative Änderung der Energie a), Impuls b) am Ende der Systemsimulation N=512 Körper nach verschiedenen Methoden in Abhängigkeit von der Anzahl der Funktionsberechnungen fn mit vierfacher Präzision (__float128) |
Die Abbildungen 3a und 3b zeigen, dass die Verwendung von Berechnungen mit vierfacher Genauigkeit die relativen Energieverluste bis zu reduzieren kann
10−23 Sie müssen jedoch verstehen, dass die Rechenzeit im Vergleich zur doppelten Genauigkeit um zwei Größenordnungen erhöht ist. Wie bei Berechnungen mit doppelter Genauigkeit das beste Verhältnis von Genauigkeit und Anzahl der Funktionsberechnungen
fn besitzen Methoden von Adams und Dorman-Prince fünfter Ordnung.
Die Dorman-Prince- und Werner-
Methoden gehören zur Klasse der
verschachtelten Methoden und können gleichzeitig zwei Lösungen mit hoher und niedriger Genauigkeit berechnen (für die Dorman-Prince-Methode die Ordnungen 5 und 4 und für die Werner-Methode die Ordnungen 6 und 5). Wenn diese beiden Lösungen sehr unterschiedlich sind, können wir den aktuellen Integrationsschritt in kleinere aufteilen. Dadurch können wir den Integrationsschritt dynamisch ändern und nur in den Bereichen reduzieren, in denen er erforderlich ist.
Vergleichen wir die Dorman-Prince-, Werner- und Adams-Methoden fünfter Ordnung detaillierter über ein längeres Simulationsintervall unseres Systems (
Tmax=300 )

|
a) Die relative Änderung von Energie, Impuls und deren Moment im Modellierungsprozess
|

|
b) Anzahl der Funktionsberechnungen fn auf das Simulationsintervall DeltaT=0,3
|
Abb. 4. Abhängigkeiten der relativen Änderung der physikalischen Parameter des Systems und der Anzahl der Funktionsberechnungen fn von Zeit zu Zeit Modellierung mit der Dorman-Prince-Methode mit variablen Schritten h=10−5 dots10−9
|

|
a) Die relative Änderung von Energie, Impuls und deren Moment im Modellierungsprozess
|

|
b) Anzahl der Funktionsberechnungen fn auf das Simulationsintervall DeltaT=0,3
|
Abb. 5. Abhängigkeiten der relativen Änderung der physikalischen Parameter des Systems und der Anzahl der Funktionsberechnungen fn versus Zeitmodellierung nach der Werner-Methode mit variablem Schritt h=10−5 dots10−9
|

|
Abb. 6. Relative Änderung von Energie, Impuls und Moment im Modellierungsprozess nach der Adams-Bashfort-Methode fünfter Ordnung mit einem Schritt h=10−6
|

|
Abb. 7. Abhängigkeiten von der Anzahl der Funktionsberechnungen fn für Adams-, Dorman-Prince- und Werner-Methoden fünfter Ordnung aus der Simulationszeit
|
Es ist ersichtlich, dass in der Anfangsphase der Entwicklung unseres Systems (
T<25 ) Alle drei Methoden weisen ähnliche Eigenschaften auf, aber in späteren Stadien treten bestimmte Ereignisse im System auf, wodurch Fehler in den Hauptparametern des Systems (Gesamtenergie, Impuls und Impuls) stark springen. Die Methoden von Dorman-Prince und Werner bewältigen diese Änderungen jedoch viel besser, da der Integrationsschritt in „komplexen“ Abschnitten reduziert werden kann, wodurch die Anzahl der Funktionsberechnungen zunimmt
fn wie in den 4b und 5b zu sehen, aber die Gesamtzahl der Berechnungen
fn verschachtelte Methoden sind kleiner als die Adams-Bashfort-Methode, wie in Abbildung 7 zu sehen ist.
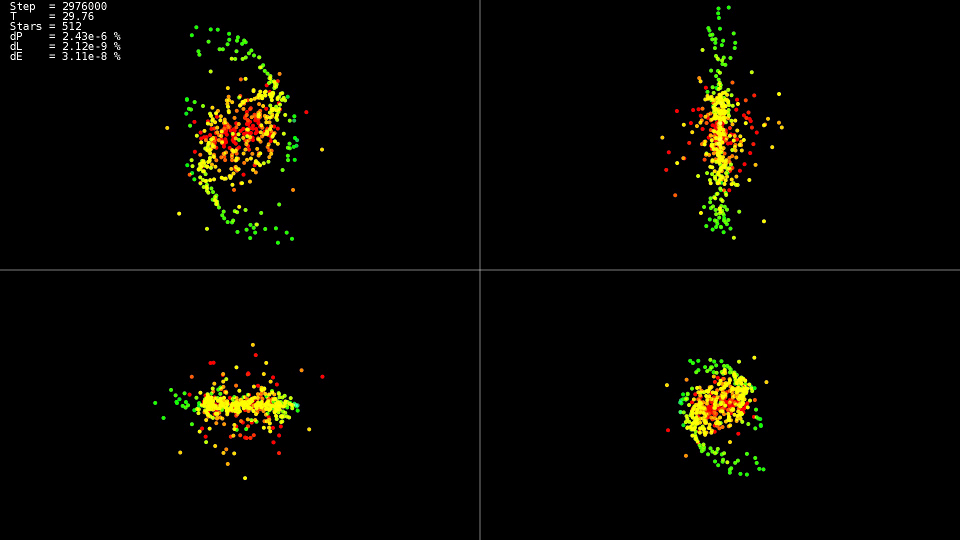
Ich frage mich, was in diesen Momenten mit dem System passiert ist
|
Video 1. Modellierung eines Systems von 512 Körpern. Dorman-Prince-Methode. Dynamischer Schritt h=10−5 dots10−9
|
Das Video zeigt das bis zum Zeitpunkt
T=25 Die Bewegung ist relativ ruhig und tritt nach einer Kollision der Zentren der „Galaxien“ auf, was zu einer starken Änderung der Flugbahnen und einem starken Anstieg der Geschwindigkeiten einiger Teilchen führt. Um die Genauigkeit der Lösung aufrechtzuerhalten, ist es außerdem erforderlich, den Integrationsschritt zu reduzieren. Verschachtelte Methoden können dies automatisch tun. Die Grafiken zeigen, dass in einigen Teilen der Entwicklung des Systems der Integrationsschritt mit um fast zwei Größenordnungen reduziert wurde
10−5 vorher
h=10−7 . Wenn wir die Adams-Methode und einen solchen Schritt über das gesamte Intervall der Entwicklung des Systems anwenden, würden wir nicht auf eine Lösung warten.
Zusammenfassung
Für die Lösung ist es besser, verschachtelte Methoden zu verwenden, mit denen Sie den Integrationsschritt dynamisch steuern und nur in "komplexen" Abschnitten der Trajektorie reduzieren können.
Verfolgen Sie nicht die Methoden höchster Ordnung. Selbst wenn der Datentyp 'double' verwendet wird, erreichen sie ihre potenziellen Fähigkeiten nicht. Durch die Verwendung der Datentypen mit größerer Genauigkeit wird die Zeit zur Lösung des Problems erheblich verlängert.
CPU-Implementierung
Nachdem die Wahl einer Methode zur Lösung von Gleichungen definiert ist, versuchen wir, die Berechnung der Wechselwirkungskraft für jedes Teilchen herauszufinden. Wir erhalten einen Doppelzyklus für alle Partikel:
Implementierungscode 'einfach'for(size_t body1 = 0; body1 < count; ++body1) { const nbvertex_t v1(rx[ body1 ], ry[ body1 ], rz[ body1 ]); nbvertex_t total_force; for(size_t body2 = 0; body2 != count; ++body2) { if(body1 == body2) { continue; } const nbvertex_t v2(rx[ body2 ], ry[ body2 ], rz[ body2 ]); const nbvertex_t force(m_data->force(v1, v2, mass[body1], mass[body2])); total_force += force; } frx[body1] = vx[body1]; fry[body1] = vy[body1]; frz[body1] = vz[body1]; fvx[body1] = total_force.x / mass[body1]; fvy[body1] = total_force.y / mass[body1]; fvz[body1] = total_force.z / mass[body1]; }
Die Gravitationskräfte für jeden Körper werden unabhängig berechnet. Um alle Prozessorkerne zu verwenden, reicht es aus, die OpenMP-Direktive vor dem ersten Zyklus zu schreiben:
Ein Stück Code aus der Implementierung von 'openmp' #pragma omp parallel for for(size_t body1 = 0; body1 < count; ++body1)
Weil Da jeder Körper mit jedem interagiert, um die Anzahl der Prozessorinteraktionen mit dem RAM zu verringern und die Cache-Nutzung zu verbessern, können wir einen Teil der Daten in den Cache laden und wiederholt verwenden:
Implementierungscode 'openmp + block' #pragma omp parallel for for(size_t n1 = 0; n1 < count; n1 += BLOCK_SIZE) { nbcoord_t x1[BLOCK_SIZE]; nbcoord_t y1[BLOCK_SIZE]; nbcoord_t z1[BLOCK_SIZE]; nbcoord_t m1[BLOCK_SIZE]; nbvertex_t total_force[BLOCK_SIZE]; for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; x1[b1] = rx[local_n1]; y1[b1] = ry[local_n1]; z1[b1] = rz[local_n1]; m1[b1] = mass[local_n1]; } for(size_t n2 = 0; n2 < count; n2 += BLOCK_SIZE) { nbcoord_t x2[BLOCK_SIZE]; nbcoord_t y2[BLOCK_SIZE]; nbcoord_t z2[BLOCK_SIZE]; nbcoord_t m2[BLOCK_SIZE]; for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { size_t local_n2 = b2 + n2; x2[b2] = rx[local_n2]; y2[b2] = ry[local_n2]; z2[b2] = rz[local_n2]; m2[b2] = mass[n2 + b2]; } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { const nbvertex_t v1(x1[ b1 ], y1[ b1 ], z1[ b1 ]); for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { const nbvertex_t v2(x2[ b2 ], y2[ b2 ], z2[ b2 ]); const nbvertex_t force(m_data->force(v1, v2, m1[b1], m2[b2])); total_force[b1] += force; } } } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; frx[local_n1] = vx[local_n1]; fry[local_n1] = vy[local_n1]; frz[local_n1] = vz[local_n1]; fvx[local_n1] = total_force[b1].x / m1[b1]; fvy[local_n1] = total_force[b1].y / m1[b1]; fvz[local_n1] = total_force[b1].z / m1[b1]; } }
Eine weitere Optimierung besteht darin, den Inhalt der Funktion zur Berechnung der Kraft im Hauptzyklus herauszunehmen und die Division und Multiplikation mit der Körpermasse m1 zu eliminieren [b1]. Neben der Tatsache, dass wir die Berechnungen ein wenig reduziert haben, kann der Compiler in einem solchen erweiterten Zyklus Vektoranweisungen des SSE- und AVX-Prozessors anwenden.
Implementierungscode 'openmp + block + optimierung' #pragma omp parallel for for(size_t n1 = 0; n1 < count; n1 += BLOCK_SIZE) { nbcoord_t x1[BLOCK_SIZE]; nbcoord_t y1[BLOCK_SIZE]; nbcoord_t z1[BLOCK_SIZE]; nbcoord_t total_force_x[BLOCK_SIZE]; nbcoord_t total_force_y[BLOCK_SIZE]; nbcoord_t total_force_z[BLOCK_SIZE]; for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; x1[b1] = rx[local_n1]; y1[b1] = ry[local_n1]; z1[b1] = rz[local_n1]; total_force_x[b1] = 0; total_force_y[b1] = 0; total_force_z[b1] = 0; } for(size_t n2 = 0; n2 < count; n2 += BLOCK_SIZE) { nbcoord_t x2[BLOCK_SIZE]; nbcoord_t y2[BLOCK_SIZE]; nbcoord_t z2[BLOCK_SIZE]; nbcoord_t m2[BLOCK_SIZE]; for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { size_t local_n2 = b2 + n2; x2[b2] = rx[local_n2]; y2[b2] = ry[local_n2]; z2[b2] = rz[local_n2]; m2[b2] = mass[n2 + b2]; } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { nbcoord_t dx = x1[b1] - x2[b2]; nbcoord_t dy = y1[b1] - y2[b2]; nbcoord_t dz = z1[b1] - z2[b2]; nbcoord_t r2(dx * dx + dy * dy + dz * dz); if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = (m2[b2]) / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; total_force_x[b1] -= dx; total_force_y[b1] -= dy; total_force_z[b1] -= dz; } } } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; frx[local_n1] = vx[local_n1]; fry[local_n1] = vy[local_n1]; frz[local_n1] = vz[local_n1]; fvx[local_n1] = total_force_x[b1]; fvy[local_n1] = total_force_y[b1]; fvz[local_n1] = total_force_z[b1]; } }
Tabelle 2. Abhängigkeit der Berechnungszeit (in Sekunden) der Funktion fn auf die Anzahl der interagierenden Körper N für verschiedene CPU-Implementierungen| N | 2048 | 4096 | 8192 | 16384 | 32768 |
|---|
| einfach | 0,0425 | 0,1651 | 0,6594 | 2,65 | 10.52 |
| openmp | 0,0078 | 0,0260 | 0,1079 | 0,417 | 1,655 |
| openmp + block + optimierung | 0,0037 | 0,0128 | 0,0495 | 0,194 | 0,774 |
Systemparameter:- System: Debian 9, Intel Core i7-5820K (6 Core)
- Compiler: gcc 6.3.0
Es ist deutlich zu sehen, dass die Version mit OpenMP-Unterstützung genau in Bezug auf die Anzahl der Kerne um das Sechsfache beschleunigt wird und die optimierte Version um etwas mehr als das Zweifache schneller ist. Bei der Optimierung sollten Sie sich also nicht nur auf die Parallelität verlassen. Interessanterweise arbeitete der Prozessor bei Einzelstromberechnungen mit einer Frequenz von 3,6 GHz, in der parallelen Version (openmp) fiel die Frequenz auf 3,4 GHz und in der parallelen und optimierten Version (openmp + Block + Optimierung) auf 3,3 GHz es hinderte sie nicht daran, 13,6-mal schneller zu arbeiten. Es ist auch ersichtlich, dass eine Zunahme der Rechenzeit mit einer Zunahme der Problemgröße quadratisch ist und eine weitere
N macht die Aufgabe in angemessener Zeit unlösbar.
GPU-Implementierung
Es besteht jedoch der Wunsch, Berechnungen noch schneller durchzuführen. Für die Beschleunigung stehen verschiedene Richtungen zur Verfügung: GPU-Berechnung, Funktionsnäherung
fn . Zunächst habe ich mich für das GPU-Computing für die OpenCL-Technologie entschieden. Für eine bequemere Arbeit wurde die
CLHPP- Bibliothek verwendet. Der Hauptvorteil von OpenCL besteht darin, dass der Code auf dem Prozessor und auf der GPU ausgeführt werden kann, was das Schreiben und Debuggen vereinfacht und die Liste der auszuführenden Hardware erweitert. Das
Oclgrind- Tool hilft beim Debuggen, das zur Laufzeit falsche OpenCL-API-Aufrufe und Speicherzugriffsprobleme anzeigt.
Opencl
Um mit OpenCL beginnen zu können, benötigen Sie eine Liste der verfügbaren Plattformen. Die gängigsten Plattformen sind AMD, Intel und NVidia.
Code std::vector<cl::Platform> platforms; cl::Platform::get(&platforms);
Als Nächstes müssen Sie nach Auswahl einer Plattform das Computergerät auswählen, das diese Plattform darstellt:
Code const cl::Platform& platform(platforms[platform_n]); std::vector<cl::Device> all_devices; platform.getDevices(CL_DEVICE_TYPE_ALL, &all_devices);
Am Ende der Vorbereitungsphase müssen ein Kontext und Warteschlangen erstellt werden, in denen Speicher zugewiesen und Berechnungen durchgeführt werden. Beispielsweise wird ein Kontext erstellt, der alle Computergeräte einer ausgewählten Plattform kombiniert:
Code zur Erstellung von Kontext und Warteschlange cl::Context context(all_devices); std::vector<cl::CommandQueue> queues; for(cl::Device device: all_devices) queues.push_back(cl::CommandQueue(context, device));
Um den Quellcode auf ein Computergerät herunterzuladen, muss er kompiliert werden, dafür ist die Klasse cl :: Program vorgesehen.
Kernel-Kompilierungscode std::vector< std::string > source_data; cl::Program::Sources sources; for(int i = 0; i != files.size(); ++i) { source_data.push_back(load_program(files[i]));
Um die Parameter einer Funktion (Kernel) zu beschreiben, die auf einem Computer ausgeführt wird, gibt es eine cl-Vorlage: make_kernel.
Beispiel für die Berechnung der Interaktionsstärke typedef cl::make_kernel< cl_int, cl_int,
Außerdem ist alles einfach: Wir deklarieren eine Variable mit dem Typ des Kernels, übergeben das kompilierte Programm und den Namen des Rechenkerns, wir können den Kernel fast wie eine normale Funktion starten.
Kernel-Startcode ComputeBlock fcompute(prog, "ComputeBlockLocal"); cl::NDRange global_range(device_data_size); cl::NDRange local_range(block_size); cl::EnqueueArgs eargs(ctx.m_queue, global_range, local_range); fcompute(eargs, ... );
Der Rechenkern für OpenCL selbst ist der Option 'openmp + block + optimierung' für die CPU sehr ähnlich, nur wird im Gegensatz zur CPU-Version der erste Zyklus mit OpenCL gesteuert (der Zyklusbereich wird durch die Variable global_range aus dem Kernel-Startcode bestimmt, und die aktuelle Iterationsnummer ist im Kernel verfügbar mit der Funktion get_global_id (0)). Zuerst wird ein Teil der Körperdaten aus dem lokalen Speicher geladen und dann verarbeitet. Der lokale Speicher ist allen Threads in der Gruppe gemeinsam, daher erfolgt der Download einmal für die Gruppe und wird von jedem Thread in der Gruppe und seitdem verarbeitet Der lokale Speicher ist viel schneller als der globale, die Berechnungen sind viel schneller.
Kernel-Code __kernel void ComputeBlockLocal(int offset_n1, int offset_n2, __global const nbcoord_t* mass, __global const nbcoord_t* y, __global nbcoord_t* f, int yoff, int foff, int points_count, int stride) { int n1 = get_global_id(0) + offset_n1; __global const nbcoord_t* rx = y + yoff; __global const nbcoord_t* ry = rx + stride; __global const nbcoord_t* rz = rx + 2 * stride; __global const nbcoord_t* vx = rx + 3 * stride; __global const nbcoord_t* vy = rx + 4 * stride; __global const nbcoord_t* vz = rx + 5 * stride; __global nbcoord_t* frx = f + foff; __global nbcoord_t* fry = frx + stride; __global nbcoord_t* frz = frx + 2 * stride; __global nbcoord_t* fvx = frx + 3 * stride; __global nbcoord_t* fvy = frx + 4 * stride; __global nbcoord_t* fvz = frx + 5 * stride; nbcoord_t x1 = rx[n1]; nbcoord_t y1 = ry[n1]; nbcoord_t z1 = rz[n1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; __local nbcoord_t x2[NBODY_DATA_BLOCK_SIZE]; __local nbcoord_t y2[NBODY_DATA_BLOCK_SIZE]; __local nbcoord_t z2[NBODY_DATA_BLOCK_SIZE]; __local nbcoord_t m2[NBODY_DATA_BLOCK_SIZE];
Cuda
Die Implementierung für die NVidia CUDA-Plattform ist etwas einfacher als für OpenCL. Wir müssen den Gerätekontext nicht erstellen und die Ausführungswarteschlange selbst verwalten (zumindest bis wir eine Implementierung mit mehreren GPUs durchführen möchten). Wie im Fall von OpenCL müssen wir Speicher auf der GPU zuweisen, unsere Daten in diese kopieren und dann können wir den Rechenkern starten.
Weitere Informationen zur Arbeit mit CUDA finden Sie
hier .
CUDA-Kernel-Startcode dim3 grid(count / block_size); dim3 block(block_size); size_t shared_size(4 * sizeof(nbcoord_t) * block_size); kfcompute <<< grid, block, shared_size >>> (... ...);
Im Gegensatz zu OpenCL gibt CUDA nicht den gesamten Bereich der Iterationen an (in der OpenCL-Implementierung ist es global_range), sondern legt die Rastergröße und die Blockgrößen im Raster fest. Dementsprechend ändert sich die Berechnung der aktuellen Körpernummer geringfügig, andernfalls ist der Kernel OpenCL mit Ausnahme anderer Namen sehr ähnlich Synchronisations- und Spezifiziererfunktionen für den gemeinsam genutzten Speicher. Ein weiteres nützliches Unterscheidungsmerkmal von CUDA ist, dass wir die erforderliche Größe des gemeinsam genutzten Speichers angeben können, wenn der Kernel gestartet wird. Wie in der OpenCL-Implementierung kopieren wir zu Beginn jedes Iterationsblocks einen Teil der Daten in den gemeinsam genutzten Speicher und arbeiten dann mit diesem Speicher aus allen Threads des Blocks.
CUDA-Kernelcode __global__ void kfcompute(int offset_n2, const nbcoord_t* y, int yoff, nbcoord_t* f, int foff, const nbcoord_t* mass, int points_count, int stride) { int n1 = blockDim.x * blockIdx.x + threadIdx.x; const nbcoord_t* rx = y + yoff; const nbcoord_t* ry = rx + stride; const nbcoord_t* rz = rx + 2 * stride; nbcoord_t x1 = rx[n1]; nbcoord_t y1 = ry[n1]; nbcoord_t z1 = rz[n1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; extern __shared__ nbcoord_t shared_xyzm_buf[]; nbcoord_t* x2 = shared_xyzm_buf; nbcoord_t* y2 = x2 + blockDim.x; nbcoord_t* z2 = y2 + blockDim.x; nbcoord_t* m2 = z2 + blockDim.x; for(int b2 = 0; b2 < points_count; b2 += blockDim.x) { int n2 = b2 + offset_n2 + threadIdx.x;
Tabelle 3. Die Abhängigkeit der Berechnungszeit (in Sekunden) fn auf die Anzahl der interagierenden Körper N für verschiedene GPU-Implementierungen und bessere CPU-Implementierungen| N | 4096 | 8192 | 16384 | 32768 | 65536 | 131072 |
|---|
| openmp + block + optimierung | 0,0128 | 0,0495 | 0,194 | 0,774 | --- ---. | --- ---. |
| OpenCL + halbe NVidia K80 | 0,004 | 0,008 | 0,026 | 0,134 | 0,322 | 1.18 |
| CUDA + halbe NVidia K80 | 0,004 | 0,008 | 0,0245 | 0,115 | 0,291 | 1.13 |
Wo bekomme ich NVidia K80?OpenCL- und CUDA-Implementierungen wurden mit dem kostenlosen
Colab- Dienst von Google gestartet, der den Zugriff auf NVidia K80-Computer ermöglicht. Weitere Informationen zum Arbeiten mit diesem Dienst auf dem
Hub finden Sie hier Im Allgemeinen ist das Ergebnis enttäuschend, nur 5-6 mal schneller als die CPU-Implementierung. Selbst wenn wir Berechnungen für den gesamten K80 durchführen, werden wir bis zu 12-mal beschleunigen, aber seitdem Da die Komplexität der Aufgabe quadratisch ist, können wir in angemessener Zeit nicht 32768 interagierende Körper verarbeiten, sondern 131072, was nur viermal mehr ist.
Funktionsannäherung fn
Wenn Sie sich die Funktion genau ansehen, die durch die Anziehungskraft zweier Körper festgelegt wird, ist klar, dass sie mit der Entfernung quadratisch abnimmt. Daher können wir die Wechselwirkungskraft zwischen nahen Körpern und ungefähr zwischen entfernten Körpern genau berechnen. Ein berühmter Ansatz
ist ein von D. Barnes und P. Hat vorgeschlagener
Treecode- Algorithmus. Im simulierten Raum wird ein
Octree-Baum konstruiert, der in seinen Blättern die Koordinaten und Massen der simulierten Körper enthält. Die übergeordneten Knoten enthalten den Schwerpunkt, die Gesamtmasse der untergeordneten Knoten und den Radius der Kugel, die um die Körper der untergeordneten Knoten beschrieben wird. Die Wurzel des Baumes enthält den Massenschwerpunkt aller Körper, ihre Gesamtmasse und den Radius der um sie herum beschriebenen Kugel. Bei der Berechnung der Wechselwirkungskraft wird zunächst der Abstand zur Wurzel des Baums berücksichtigt, wenn das Verhältnis des Abstands zum Knoten zu seinem Radius größer als eine Konstante ist
lambdakrit Dann wird die Wurzel als ein Körper mit Koordinaten betrachtet, die den Koordinaten des Massenschwerpunkts der darin enthaltenen Körper entsprechen, und als eine Masse, die der Summe der Massen der Tochterknoten entspricht, wenn das Verhältnis kleiner oder gleich ist
lambdakrit Dann wird die Prozedur für jeden untergeordneten Knoten rekursiv wiederholt. Wenn ein Blatt eines Baumes erreicht ist, wird die Wechselwirkungskraft auf die übliche Weise berücksichtigt. Wenn also ein Körper weit von der kompakten Gruppe anderer Körper entfernt ist, wird diese Gruppe ihm als ein Körper dargestellt, und die Wechselwirkungskraft wird nicht bis zu jedem Körper berechnet, sondern nur bis zu einem Körper. Aufgrund dessen nimmt die Komplexität des Algorithmus mit ab
O(N2) vorher
O(N log(N)) auf Kosten eines gewissen Genauigkeitsverlustes.
Bei meinem Ansatz habe ich mich entschieden, nicht den
Octree- Baum , sondern den
kd-Baum zu verwenden, weil Es ist einfacher zu verwenden und hat im Vergleich zum Octree einen geringeren Speicheraufwand.
Kehren wir zur Implementierung auf der CPU zurück. Ein kd-Baumknoten kann in Form einer Klasse dargestellt werden, die Zeiger auf die linken und rechten Nachkommen und Informationen zu den Koordinaten und der Masse enthält:
Kd-Baumknoten class node { node* m_left;
Mit dieser Methode zum Speichern des Baums haben wir zwei Möglichkeiten, den Baum zu durchlaufen: entweder explizite Rekursion verwenden oder den Stapel selbst verwenden. Ich entschied mich für die zweite Option.
Berechnung der Wechselwirkungskraft durch Überqueren eines Baumes nbvertex_t force_compute(const nbvertex_t& v1, const nbcoord_t mass1) { nbvertex_t total_force; node* stack_data[MAX_STACK_SIZE] = {}; node** stack = stack_data; node** stack_head = stack; *stack++ = m_root; while(stack != stack_head) { node* curr = *--stack; const nbcoord_t distance_sqr((v1 - curr->m_mass_center).norm()); if(distance_sqr > curr->m_radius_sqr) {
Wie bei der „exakten“ CPU-Implementierung wird die Kraftberechnungsfunktion für jeden Körper aufgerufen. Der Zyklus über alle Körper hinweg kann mithilfe von OpenMP-Anweisungen einfach parallelisiert werden.
In diesem Fall beziehen sich benachbarte Schleifeniterationen jedoch auf völlig unterschiedliche Teile des Baums, was eine effiziente Nutzung des Prozessor-Cache nicht ermöglicht. Um dieses Problem zu lösen, müssen alle Körper nicht in der ursprünglichen Reihenfolge durchlaufen werden, sondern in der Reihenfolge, in der sich die Körper in den Blättern des kd-Baums befinden. Dann treten benachbarte Iterationen für Körper auf, die sich im Raum befinden, und durchlaufen den Baum auf fast denselben Pfaden.
Baumblattdurchquerung template<class Visitor> void traverse(Visitor visit) { node* stack_data[MAX_STACK_SIZE] = {}; node** stack = stack_data; node** stack_head = stack; *stack++ = m_root; while(stack != stack_head) { node* curr = *--stack; if(curr->m_radius_sqr > 0) {
Diese Implementierung hat ein weiteres Problem: Es gibt keine universelle Möglichkeit, eine solche Baumdurchquerung zu parallelisieren. Aber wir können die Art und Weise, wie der Baum im Speicher gespeichert wird, vollständig ändern - wir können alle Knoten in einem linearen Array speichern und die Speicherung von Zeigern auf Nachkommen vollständig aufgeben, analog zur Konstruktion eines
binären Heaps . Beim Starten der Nummerierung von Knoten mit
1 wenn sich der Knoten in der Zellennummer befindet
i , dann ist sein linkes Kind in der Zelle
2i , rechtes Kind in der Zelle
2 i + 1 und das übergeordnete Element in der Zelle
i / 2 . Der rechte Knoten entspricht dem linken mit der Nummer
ich wird eine Nummer haben
i + 1 . Das Array selbst hat eine Länge
2 N. und alle Blattknoten befinden sich im letzten
N. Zellen, außerdem werden eng beabstandete Knoten in engen Zellen des Arrays lokalisiert. Die Tree Traversal-Funktion ändert sich ein wenig, aber das Framework bleibt gleich:
Berechnung der Kraft durch Überqueren eines Baums in einem Array nbvertex_t force_compute(const nbvertex_t& v1, const nbcoord_t mass1) { nbvertex_t total_force; size_t stack_data[MAX_STACK_SIZE] = {}; size_t* stack = stack_data; size_t* stack_head = stack; *stack++ = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { size_t curr = *--stack; const nbcoord_t distance_sqr((v1 - m_mass_center[curr]).norm()); if(distance_sqr > m_radius_sqr[curr]) { total_force += force(v1, m_mass_center[curr], mass1, m_mass[curr]); } else { size_t left(left_idx(curr)); size_t rght(rght_idx(curr)); if(rght < m_body_n.size()) { *stack++ = rght; } if(left < m_body_n.size()) { *stack++ = left; } } } return total_force; }
Dies sind jedoch nicht alle Möglichkeiten, die uns das Speichern der Knoten im Array eröffnet - wir können den Stapel beim Crawlen ablehnen. Dazu fügen wir in dem Code-Zweig, in dem wir zu den untergeordneten Knoten des Knotens gehen, die Funktion zur Berechnung des nächsten Knotens hinzu (
n e x t u p ) und in dem Zweig, in dem wir die Wechselwirkungskraft berechnen, fügen wir die Berechnung des nächsten Knotens hinzu, wobei der aktuelle Teilbaum übersprungen wird (
s k i p d o w n )
Um den aktuellen Teilbaum zu überspringen, müssen wir zur Wurzel (Richtung) gehen
D. ), während wir uns im rechten Nachkommen befinden, gehen wir, sobald wir den linken erreichen, zum entsprechenden rechten Teilbaum (Richtung)
R. ), wenn wir zur Wurzel kommen, ist die Baumdurchquerung abgeschlossen.
Funktionscode des Teilbaums überspringen index_t skip_down(index_t idx) {
 Abbildung 8. Überspringen eines Teilbaums s k i p d o w n .
Abbildung 8. Überspringen eines Teilbaums s k i p d o w n .Um zum nächsten Knoten zu gelangen, gehen Sie nach Möglichkeit zum linken Kind (Richtung)
U. ), und wenn es keinen Nachkommen gibt, gehen Sie mit der Funktion zum nächsten Knoten 'von unten'
s k i p d o w n .
Gehen Sie zum nächsten Knotenfunktionscode index_t next_up(index_t idx, index_t tree_size) { index_t left = left_idx(idx); if(left < tree_size) { return left; } return skip_down(idx); }
 Abbildung 9. Übergänge zum nächsten Knoten n e x t u p .
Abbildung 9. Übergänge zum nächsten Knoten n e x t u p .Es scheint, dass wir die Rekursion durch eine Schleife ersetzt haben
w ä h r e n d in Funktion
s k i p d o w n , und ein solcher Ersatz gibt nichts, aber lassen Sie uns sehen, wie man feststellt, ob ein Knoten mit einer Nummer
ich rechter Nachkomme. Dies kann einfach durch Überprüfen der ungeraden Zahl erfolgen (das richtige Kind befindet sich in der Zelle
2 i + 1 ), dafür reicht es zu berechnen
i \ & 1 . Das heißt, Wir teilen die Zahl
i zwei, wenn das niedrigstwertige Bit auf eins gesetzt ist. Dies kann jedoch ohne Schleife erfolgen. In vielen Prozessoren gibt es einen Befehl zum
Suchen des ersten Satzes , der die Position des ersten gesetzten Bits zurückgibt. Mit ihm wird die Schleife in vier Prozessorbefehle umgewandelt.
Optimierter Teilbaum-Funktionscode überspringen index_t skip_down(index_t idx) { idx = idx >> (__builtin_ffs(~idx) - 1); return left2right(idx); }
Danach können wir den Stapel von der Baumdurchlauffunktion ausschließen und durch ein Paar ersetzen
skipdown+nextup danach sieht die Funktion noch einfacher aus:
Erzwingen Sie die Berechnung, indem Sie einen Baum in einem Array ohne Verwendung eines Stapels durchlaufen nbvertex_t force_compute(const nbvertex_t& v1, const nbcoord_t mass1) const { nbvertex_t total_force; size_t curr = NBODY_HEAP_ROOT_INDEX; size_t tree_size = m_mass_center.size(); do { const nbcoord_t distance_sqr((v1 - m_mass_center[curr]).norm()); if(distance_sqr > m_radius_sqr[curr]) { total_force += force(v1, m_mass_center[curr], mass1, m_mass[curr]); curr = skip_down(curr); } else { curr = next_up(curr, tree_size); } } while(curr != NBODY_HEAP_ROOT_INDEX); return total_force; }
Insgesamt haben wir sechs Kombinationen aus Baumdurchquerung und Kraftberechnung erhalten. Vergleichen Sie diese Ansätze hinsichtlich Rechenzeit und Qualität. Als Qualitätsmaß nehmen wir die relative Änderung der Gesamtenergie des Systems nach 100 Iterationen. Als Modellsystem nehmen wir zwei interagierende „Galaxien“, bestehend aus16384Körper jeweils.Tabelle 4. Kombinationen aus Baumdurchquerungsmethode und Kraftberechnung| Baumdurchquerungs- / Kraftberechnungstyp | | '' | '' |
|---|
| cycle+tree | cycle+heap | cycle+heapstackless |
|---|
| nestedtree+tree | nestedtree+heap | nestedtree+heapstackless |
|---|

| 
|
| a) fn ( λcrit ) | b) ( λcrit ) |
| 10. fn CPU ( N=32768 ) |
Es ist zu sehen, dass die Implementierung von 'nestedtree + tree' hoffnungslos in der Geschwindigkeit zurückliegt, weil es fehlt die Parallelität. Implementierungen mit der Position von Baumknoten im Array und der Indizierung wie in einem binären Heap liegen an der Spitze. Die relative Energieänderung ist für alle Varianten mit vernachlässigbarλcrit>1 .
Auch in Abb. 10a zeigt, dass für (λcrit<10 ) alle Varianten der Funktionsberechnung fn Die optimierte Version der exakten Berechnung ('openmp + block + optimierung') wurde mit einer weiteren Steigerung deutlich schneller λcrit Implementierungen mit einem Baum verlieren die genaue Version.GPU-Baumdurchquerung
Ich habe versucht, den Baum auf der GPU sowohl mit OpenCL-Technologie als auch mit CUDA zu umgehen. Die Option zum Speichern von Knoten in Form eines Baums wurde sofort verworfen, und es blieben nur Optionen zum Speichern des Baums in einem Array mit Indizierung wie in einem binären Heap. Im Allgemeinen unterscheidet sich die Implementierung des Computerkerns nicht wesentlich von der CPU-Version.OpenCL-Kernel zur Berechnung der Stärke durch Durchlaufen eines Baums (Durchlaufen in der Reihenfolge der Nummerierungskörper) __kernel void ComputeTreeBH(int offset_n1, int points_count, int tree_size, __global const nbcoord_t* y, __global nbcoord_t* f, __global const nbcoord_t* tree_cmx, __global const nbcoord_t* tree_cmy, __global const nbcoord_t* tree_cmz, __global const nbcoord_t* tree_mass, __global const nbcoord_t* tree_crit_r2) { int n1 = get_global_id(0) + offset_n1; int stride = points_count; __global const nbcoord_t* rx = y; __global const nbcoord_t* ry = rx + stride; __global const nbcoord_t* rz = rx + 2 * stride; __global const nbcoord_t* vx = rx + 3 * stride; __global const nbcoord_t* vy = rx + 4 * stride; __global const nbcoord_t* vz = rx + 5 * stride; __global nbcoord_t* frx = f; __global nbcoord_t* fry = frx + stride; __global nbcoord_t* frz = frx + 2 * stride; __global nbcoord_t* fvx = frx + 3 * stride; __global nbcoord_t* fvy = frx + 4 * stride; __global nbcoord_t* fvz = frx + 5 * stride; nbcoord_t x1 = rx[n1]; nbcoord_t y1 = ry[n1]; nbcoord_t z1 = rz[n1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int stack_data[MAX_STACK_SIZE] = {}; int stack = 0; int stack_head = stack; stack_data[stack++] = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { int curr = stack_data[--stack]; nbcoord_t dx = x1 - tree_cmx[curr]; nbcoord_t dy = y1 - tree_cmy[curr]; nbcoord_t dz = z1 - tree_cmz[curr]; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > tree_crit_r2[curr]) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tree_mass[curr] / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; } else { int left = left_idx(curr); int rght = rght_idx(curr); if(left < tree_size) { stack_data[stack++] = left; } if(rght < tree_size) { stack_data[stack++] = rght; } } } frx[n1] = vx[n1]; fry[n1] = vy[n1]; frz[n1] = vz[n1]; fvx[n1] = res_x; fvy[n1] = res_y; fvz[n1] = res_z; }
In der ersten Version begann die Baumdurchquerung in der Reihenfolge der Nummerierung der Körper im ursprünglichen Array, sodass benachbarte Threads völlig unterschiedliche Teile des Baums umgingen, was sich negativ auf die Leistung des GPU-Speichercaches auswirkte. Daher wurde in der zweiten Ausführungsform eine Durchquerung ausgehend von der Spitze des Baums angewendet, in diesem Fall beginnen benachbarte Fäden, den Baum entlang des gleichen Pfades zu durchqueren, weil benachbarte Baumwipfel sind in der Nähe und im Weltraum. Es ist auch wichtig, dass wir die Nummerierung im Array der Baumknoten nicht von Grund auf neu gewählt haben, sondern von einem. In diesem Fall werden die Blätter des Baums in der zweiten Hälfte des Arrays gespeichert. Wenn die Anzahl der Körper gleich der Zweierpotenz ist, haben wir über den Index tn1 den gleichen Zugriff auf den Speicher.OpenCL-Kernel zur Berechnung der Stärke durch Durchlaufen eines Baums (Durchlaufen der Nummerierungsknoten eines Baums) __kernel void ComputeHeapBH(int offset_n1, int points_count, int tree_size, __global const nbcoord_t* y, __global nbcoord_t* f, __global const nbcoord_t* tree_cmx, __global const nbcoord_t* tree_cmy, __global const nbcoord_t* tree_cmz, __global const nbcoord_t* tree_mass, __global const nbcoord_t* tree_crit_r2, __global const int* body_n) { int tree_offset = points_count - 1 + NBODY_HEAP_ROOT_INDEX; int stride = points_count; int tn1 = get_global_id(0) + offset_n1 + tree_offset; int n1 = body_n[tn1]; nbcoord_t x1 = tree_cmx[tn1]; nbcoord_t y1 = tree_cmy[tn1]; nbcoord_t z1 = tree_cmz[tn1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int stack_data[MAX_STACK_SIZE] = {}; int stack = 0; int stack_head = stack; stack_data[stack++] = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { int curr = stack_data[--stack]; nbcoord_t dx = x1 - tree_cmx[curr]; nbcoord_t dy = y1 - tree_cmy[curr]; nbcoord_t dz = z1 - tree_cmz[curr]; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > tree_crit_r2[curr]) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tree_mass[curr] / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; } else { int left = left_idx(curr); int rght = rght_idx(curr); if(left < tree_size) { stack_data[stack++] = left; } if(rght < tree_size) { stack_data[stack++] = rght; } } } __global const nbcoord_t* vx = y + 3 * stride; __global const nbcoord_t* vy = y + 4 * stride; __global const nbcoord_t* vz = y + 5 * stride; __global nbcoord_t* frx = f; __global nbcoord_t* fry = frx + stride; __global nbcoord_t* frz = frx + 2 * stride; __global nbcoord_t* fvx = frx + 3 * stride; __global nbcoord_t* fvy = frx + 4 * stride; __global nbcoord_t* fvz = frx + 5 * stride; frx[n1] = vx[n1]; fry[n1] = vy[n1]; frz[n1] = vz[n1]; fvx[n1] = res_x; fvy[n1] = res_y; fvz[n1] = res_z; }
Beim Durchlaufen der Nummerierungsreihenfolge von Baumknoten wurde eine Leistungssteigerung erzielt. Diese Option kann aber auch verbessert werden. Der globale Speicher, in dem sich die Baumknoten derzeit befinden, ist für den gemeinsamen Zugriff optimiert , d. H. Threads einer Gruppe sollten Wörter lesen, die sich in einem Speicherblock befinden. In unserem Fall beginnt die Baumdurchquerung auf denselben Pfaden, und wir fordern dieselben Daten mit allen Threads der Gruppe an. Je weiter wir jedoch tiefer in den Baum vordringen, desto mehr divergieren die Pfade benachbarter Threads, und wir müssen unterschiedliche Daten anfordern, was die Leistung des Subsystems mehrmals verringert Speicher. Die Knoten jedes Teilbaums, der zu derselben Ebene gehört, befinden sich jedoch in relativ engen Speicherzellen. Das heißt,
Beim Durchlaufen des restlichen Baums greifen die benachbarten Threads des Rechenkerns nicht auf dieselben Knoten des Baums zu, sondern befinden sich eng im Speicher. Um diesen Speicherzugriff zu optimieren, kann ein Texturspeicher verwendet werden. Aber es gibt einen Haken. Im Moment unterstützen Texturen das Arbeiten mit Daten mit doppelter Genauigkeit nicht (wir möchten genau berechnen). In CUDA gibt es jedoch eine __hiloint2double- Funktion , die eine Zahl mit doppelter Genauigkeit aus zwei Ganzzahlen sammelt.Zahlenanforderungscode mit doppelter Genauigkeit aus einer Textur, in der Ganzzahlen gespeichert sind template<> struct nb1Dfetch<double> { typedef double4 vec4; static __device__ double fetch(cudaTextureObject_t tex, int i) { int2 p(tex1Dfetch<int2>(tex, i)); return __hiloint2double(py, px); } static __device__ vec4 fetch4(cudaTextureObject_t tex, int i) { int ii(2 * i); int4 p1(tex1Dfetch<int4>(tex, ii)); int4 p2(tex1Dfetch<int4>(tex, ii + 1)); vec4 d4 = {__hiloint2double(p1.y, p1.x), __hiloint2double(p1.w, p1.z), __hiloint2double(p2.y, p2.x), __hiloint2double(p2.w, p2.z) }; return d4; } };
In diesem Fall wurden zwei Implementierungen durchgeführt, in denen jedes Element des Baums (x, y, z, tree_crit_r2) unabhängig voneinander angefordert wurde, und in der zweiten Implementierung wurden diese Anforderungen kombiniert. Die Anforderung für die Masse des Knotens tritt viel seltener auf, nur wenn die Bedingung r2> tree_crit_r2 [curr] erfüllt ist. Daher ist es nicht sinnvoll, diese Anforderung mit den anderen zu kombinieren. Ein weiteres nützliches Merkmal des CUDA-Frameworks ist die Möglichkeit, das Verhältnis der Größe des L1-Cache zur Größe des gemeinsam genutzten Speichers ( cudaFuncSetCacheConfig ) zu steuern . Im Falle einer Baumdurchquerung verwenden wir keinen gemeinsam genutzten Speicher, sodass wir den L1-Cache zum Nachteil erhöhen können.CUDA-Kernel zur Berechnung der Stärke durch Durchlaufen eines Baums (Durchlaufen der Nummerierungsknoten eines Baums) __global__ void kfcompute_heap_bh_tex(int offset_n1, int points_count, int tree_size, nbcoord_t* f, cudaTextureObject_t tree_xyzr, cudaTextureObject_t tree_mass, const int* body_n) { nb1Dfetch<nbcoord_t> tex; int tree_offset = points_count - 1 + NBODY_HEAP_ROOT_INDEX; int stride = points_count; int tn1 = blockDim.x * blockIdx.x + threadIdx.x + offset_n1 + tree_offset; int n1 = body_n[tn1]; nbvec4_t xyzr = tex.fetch4(tree_xyzr, tn1); nbcoord_t x1 = xyzr.x; nbcoord_t y1 = xyzr.y; nbcoord_t z1 = xyzr.z; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int stack_data[MAX_STACK_SIZE] = {}; int stack = 0; int stack_head = stack; stack_data[stack++] = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { int curr = stack_data[--stack]; nbvec4_t xyzr2 = tex.fetch4(tree_xyzr, curr); nbcoord_t dx = x1 - xyzr2.x; nbcoord_t dy = y1 - xyzr2.y; nbcoord_t dz = z1 - xyzr2.z; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > xyzr2.w) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tex.fetch(tree_mass, curr) / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; } else { int left = nbody_heap_func<int>::left_idx(curr); int rght = nbody_heap_func<int>::rght_idx(curr); if(left < tree_size) { stack_data[stack++] = left; } if(rght < tree_size) { stack_data[stack++] = rght; } } } f[n1 + 3 * stride] = res_x; f[n1 + 4 * stride] = res_y; f[n1 + 5 * stride] = res_z; }
Eine Analyse des Programms im nvprof-Profiler ergab, dass selbst bei Verwendung des Texturspeichers zum Speichern eines Baums der globale Speicher immer noch sehr stark belastet ist.In der Tat wird in CUDA der gesamte Kernelspeicher, der an "berechnete" Adressen adressiert ist, im globalen Speicher gespeichert, und dementsprechend befindet sich der Stapel, der zum Durchlaufen des Baums benötigt wird, im globalen Speicher und verschlingt einen erheblichen Teil der Speicherchipbandbreite, da der Stapel über einen Speicher verfügt Jeder Run-Thread und es gibt viele Threads.Glücklicherweise wissen wir bereits, wie man einen Baum ohne Verwendung eines Stapels durchquert. Ergänzend zum vorherigen Rechenkern mit den Funktionen zur Berechnung des nächsten Baumknotens erhalten wir einen neuen Kernel, der darüber hinaus kompakter ist.CUDA-Kern zur Berechnung der Stärke durch Durchlaufen eines Baums ohne Verwendung eines Stapels __global__ void kfcompute_heap_bh_stackless(int offset_n1, int points_count, int tree_size, nbcoord_t* f, cudaTextureObject_t tree_xyzr, cudaTextureObject_t tree_mass, const int* body_n) { nb1Dfetch<nbcoord_t> tex; int tree_offset = points_count - 1 + NBODY_HEAP_ROOT_INDEX; int stride = points_count; int tn1 = blockDim.x * blockIdx.x + threadIdx.x + offset_n1 + tree_offset; int n1 = body_n[tn1]; nbvec4_t xyzr = tex.fetch4(tree_xyzr, tn1); nbcoord_t x1 = xyzr.x; nbcoord_t y1 = xyzr.y; nbcoord_t z1 = xyzr.z; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int curr = NBODY_HEAP_ROOT_INDEX; do { nbvec4_t xyzr2 = tex.fetch4(tree_xyzr, curr); nbcoord_t dx = x1 - xyzr2.x; nbcoord_t dy = y1 - xyzr2.y; nbcoord_t dz = z1 - xyzr2.z; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > xyzr2.w) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tex.fetch(tree_mass, curr) / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; curr = nbody_heap_func<int>::skip_idx(curr); } else { curr = nbody_heap_func<int>::next_up(curr, tree_size); } } while(curr != NBODY_HEAP_ROOT_INDEX); f[n1 + 3 * stride] = res_x; f[n1 + 4 * stride] = res_y; f[n1 + 5 * stride] = res_z; }
Die Leistung der auf der GPU ausgeführten Kerne hängt stark von der Größe der Blöcke ab, in die wir die Aufgabe unterteilen. Abhängig von dieser Größe, wie viele Register, lokaler Speicher und andere Ressourcen für jeden Computer-Thread verfügbar sind. Sie müssen auch berücksichtigen, dass während des Wartens auf den Speicherzugriff in einem Thread ein anderer Thread Berechnungen auf dem Shader-Prozessor ausführen kann. Bei einer ausreichenden Anzahl gleichzeitig ausgeführter Threads auf einem Prozessor wird die Speicherzugriffszeit hinter den Berechnungen verborgen. Bevor wir die Leistung unserer Kerne vergleichen, müssen wir daher für jeden die optimale Blockgröße berechnen. Lassen Sie uns einen Vergleich der Hälfte von NVidia K80 anstellen.Tabelle 5. Die Abhängigkeit der Berechnungszeit (in Sekunden) fn GPU N=131072 und λcrit=10| / | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 1024 |
|---|
| opencl+dense | 5.77 | 2.84 | 1.46 | 1.13 | 1.15 | 1.14 | 1.14 | 1.13 |
| cuda+dense | 5.44 | 2.55 | 1.27 | 0.96 | 0.97 | 0.99 | 0.99 | - - |
| opencl+heap+cycle | 5.88 | 5.65 | 5.74 | 5.96 | 5.37 | 5.38 | 5.35 | 5.38 |
| opencl+heap+nested | 4.54 | 3.68 | 3.98 | 5.25 | 5.46 | 5.41 | 5.48 | 5.31 |
| cuda+heap+nested | 3.62 | 2.81 | 2.68 | 4.26 | 4.84 | 4,75 | 4.8 | 4.67 |
| cuda+heap+nested+tex | 2.6 | 1.51 | 0.912 | 0.7 | 1,85 | 1.75 | 1,69 | 1.61 |
| cuda+heap+nested+tex+stackless | 2.3 | 1.29 | 0.773 | 0,5 | 0,51 | 0,52 | 0,52 | 0,52 |
6. ( ) fn GPU N=1M und λcrit=4| / | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 1024 |
|---|
| opencl+dense | 366 | 179 | 89.9 | 69.3 | 70.3 | 69.1 | 68.9 | 68.0 |
| cuda+dense | 346 | 162 | 79.6 | 60.8 | 60.8 | 60.7 | 59.6 | - - |
| opencl+heap+cycle | 16.2 | 18.2 | 20.1 | 21.2 | 21.2 | 21.3 | 21.2 | 21.1 |
| opencl+heap+nested | 10.5 | 7.63 | 6.38 | 8.23 | 9.95 | 9.89 | 9.65 | 9.66 |
| cuda+heap+nested | 8.67 | 6.44 | 5.39 | 5.93 | 8.65 | 8.61 | 8.41 | 8.27 |
| cuda+heap+nested+tex | 6.38 | 3.57 | 2.13 | 1.44 | 3.56 | 3.46 | 3.30 | 3.29 |
| cuda+heap+nested+tex+stackless | 5.78 | 3.19 | 1.83 | 1.21 | 1.11 | 1.10 | 1.11 | 1.13 |
Eine schwierige Situation, aber im Gegensatz zur CPU-Version der Baumdurchquerung ist klar, dass jeder Optimierungsschritt greifbare Ergebnisse bringt. Die Implementierung von 'opencl + heap + cycle' ist fast sechsmal langsamer als eine exakte Lösung mit Vollfunktionsberechnungfn .
Eine Implementierung von 'opencl + heap + nested', bei der eine Baumdurchquerung von benachbarten Knoten aus gestartet wird, 1,4-mal schneller als die vorherige, weil Der Cache-Speicher wird besser genutzt. Bei der Implementierung von 'cuda + heap + nested' wurde der L1-Cache zum Nachteil des gemeinsam genutzten Speichers erhöht, wodurch die Geschwindigkeit um das 1,4-fache erhöht wurde, obwohl es möglich ist, dass bei der cuda-Implementierung der Kompilierungskernel optimaler kompiliert wird (in den Versionen von 'opencl + dens' und 'cuda' + dichte Kerne sind identisch und die Versionsleistung von cuda ist ~ 1,2-mal höher). Die signifikanteste Erhöhung der Rechengeschwindigkeit (3,8-fach) wird erreicht, wenn sich der Baum im Texturspeicher befindet und die Abfragen an die Elemente des Baumknotens kombiniert werden. Die Implementierung mit Tree Traversal ohne Verwendung des Stacks 'cuda + heap + nested + tex + stackless' ist ebenfalls 1,4-mal schneller.Darin wird die gesamte Bandbreite des Speicherbusses nur für den Zugriff auf Daten über Baumknoten verwendet und nicht für den Stapel ausgegeben. Also mitλcrit=10 Im Vergleich zur vollständigen Berechnung der Funktion konnte eine doppelte Beschleunigung erreicht werden fn . Aber
λcrit=10 Ein übermäßig großer Wert des Parameters in der Grafik der relativen Änderung der Energie im System vom Verhältnis des kritischen Abstands zum Baumknoten zu seinem Radius für die CPU-Implementierung ist ersichtlich, dass niedrigere Werte verwendet werden können λcritohne sichtbaren Genauigkeitsverlust. Versuchen wir zu variierenλcrit mit den optimalen Blockgrößen, die wir im vorherigen Schritt ermittelt haben.
| 
|
a) N=128K
| b) N=1M
|
Abb. 11. Abhängigkeit der Funktionsberechnungszeit. fn vom Verhältnis der kritischen Entfernung zum Baumknoten zu seinem Radius ( λcrit ) für verschiedene GPU-Tree-Walk-Implementierungen
|
Es ist zu sehen, dass für kleine λcrit alle Methoden der Funktionsberechnung fnGehen Sie zu den Abschlusswerten, die durch den Zeitpunkt des Erstellens des kd-Baums und der Vorbereitung der Daten für die GPU bestimmt werden. Darüber hinaus trägt die Zeit des Baumbaus wesentlich zur Gesamtzeit beiλcrit≤4, dann kann diese Zeit vernachlässigt werden. Es ist interessant festzustellen, dass wannN=128K Die Leistung verbessert sich beim Erreichen wieder λcrit=1024Dies ist höchstwahrscheinlich auf die Tatsache zurückzuführen, dass alle GPU-Threads gleichzeitig dieselben Baumscheitelpunkte durchlaufen, was die Verwendung des L1-Cache verbessert und die "Verzweigungsdivergenz" vollständig beseitigt . Es ist auch ersichtlich, dass die beste Leistung für eine Implementierung ohne Stapel (cuda + heap + verschachtelt + tex + stapellos) die Version mit dem Stapel bei etwa übertrifft1.4−1.5mal. Andere Implementierungen sind um ein Vielfaches langsamer. Um das Ergebnis zu konsolidieren, berechnen wir die Zeit auf einer GPU mit einer neueren Architektur.Ergebnisse auf GeForce GTX 1080 Ti starten (Single Accuracy)7. ( ) fn GPU N=1M und λcrit=4| / | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 1024 |
|---|
| opencl+dense | 47.8 | 23.4 | 11.6 | 11.59 | 12.8 | 12.8 | 12.8 | 12.8 |
| cuda+dense | 49.0 | 23.8 | 11.9 | 11.8 | 11.7 | 11.7 | 11.7 | 11.7 |
| opencl+heap+cycle | 7.48 | 8.26 | 7.73 | 7.36 | 7.33 | 7.27 | 7.25 | 7.26 |
| opencl+heap+nested | 1.33 | 1,20 | 1.41 | 2.46 | 2.53 | 2.49 | 2.44 | 2.47 |
| cuda+heap+nested | 1.23 | 1.10 | 1.31 | 2.28 | 2.29 | 2.28 | 2.23 | 2.25 |
| cuda+heap+nested+tex | 0.88 | 0,68 | 0.654 | 1.6 | 1.6 | 1.6 | 1.6 | 1.6 |
| cuda+heap+nested+tex+stackless | 0,71 | 0.47 | 0,45 | 0,43 | 0,43 | 0,42 | 0,41 | 0.395 |

|
12. fn ( λcrit ) GPU
|
Bei Verwendung der GeForce GTX 1080 Ti zur Berechnung beträgt der Unterschied zwischen Baumläufen mit und ohne Stapel bis zu zweimal, vorausgesetzt, wir vernachlässigen die Zeit, die zum Erstellen des Baums benötigt wird. Diese Tatsache zwingt uns, sicherzustellen, dass in Zukunft auf die GPU portiert und ein Baum erstellt wird. Ein Vergleich der Tabellen 5-7 zeigt, dass es keine einzige optimale Blockgröße für unterschiedliche Anzahlen von Körpern gibt, und noch mehr für unterschiedliche GPU-Architekturen. Darüber hinaus kann die Berechnungszeitdifferenz mehrere Male erreichen, selbst wenn Sie die Grenzwerte nicht berücksichtigen. Daher ist es vor langwierigen Berechnungen sinnvoll, die optimale Blockgröße für jede Konfiguration zu bestimmen.Die Hauptsache, die wir erreicht haben, ist die Fähigkeit, die paarweise Interaktion von etwas mehr als einer Million Körpern zu modellieren (220) für eine angemessene Zeit auf einer, nicht der neuesten GPU. Auf neueren GPUs (Tesla V100) wird es offenbar möglich sein, bereits etwa zwei Millionen interagierende Körper in angemessener Zeit zu verarbeiten, weil Ihre Leistung ist etwa viermal höher als die Hälfte des Tesla K80. Diese Zahl ist zwar auch mit der Anzahl der Sterne selbst in Zwerggalaxien wie der Kleinen Magellanschen Wolke nicht zu vergleichen , aber meiner Meinung nach beeindruckend.Fazit
Die Verwendung eingebetteter Methoden zur Lösung von Differentialgleichungen ermöglicht es, ähnliche Probleme mit einem hohen Maß an Genauigkeit zu relativ geringen Zeitkosten zu lösen, und die Verwendung von Approximationsalgorithmen zur Berechnung der paarweisen Anziehungskräfte ermöglicht es uns, kolossale Volumina interagierender Körper zu verarbeiten. Im Gegensatz zu den Außerirdischen aus den „Drei-Körper-Aufgaben“ können wir das Problem lösenN Körper und für eine kleine Anzahl von Körpern und für ganze Sternhaufen, allerdings auf Kosten eines gewissen Genauigkeitsverlustes.Visualisierung
Für diejenigen, die bis zum Ende gelesen haben, werde ich ein paar weitere Videos mit Visualisierung der Entwicklung der Körpersysteme geben.Simulation einer Kollision zweier Galaxien. Die Gesamtzahl der Leichen beträgt 60 Tausend.Modellierung der Entwicklung einer Galaxie aus einer Million Sternen. In der Mitte befindet sich ein Körper mit einer Masse von 99% der Gesamtmasse. Einzelne Partikel sind kaum zu unterscheiden. Schon eher wie eine Welle in einem Tropfen Flüssigkeit. Koloriert nach Geschwindigkeitsmodul. Niedrige Geschwindigkeit - blau, mittel - grün, hoch - rot. Es ist zu sehen, dass in der Mitte die Geschwindigkeit höher ist und allmählich bis zu den Kanten abnimmt und die niedrigste in der Äquatorialebene.Eine "dynamischere" Simulation der Entwicklung einer Millionen-Sterne-Galaxie. In der Mitte befindet sich ein Körper mit einer Masse von 10% der Gesamtmasse. Der Zentralkörper beeinflusst den Rest deutlich weniger. Zuerst fliegen die „Sterne“ auseinander, sammeln sich dann wieder zu mehreren Clustern und bilden am Ende wieder einen großen Cluster.Bei der Modellierung verließen etwa 5% der „Sterne“ unwiderruflich die ursprüngliche Region.In der 10. Sekunde ähnelt es sehr der vorhandenen Wagenradgalaxie .Den Projektcode finden Sie auf dem Github .