Was ist, wenn Sie eine Idee für ein kühles, gesundes Protein haben und es in die Realität umsetzen möchten? Möchten Sie beispielsweise einen Impfstoff gegen
H. pylori (wie das
slowenische Team auf der iGEM 2008 ) erstellen, indem Sie ein Hybridprotein erstellen, das
E. coli- Flagellinfragmente kombiniert, die die Immunantwort mit dem üblichen
H. pylori- Flagellin stimulieren?
H. pylori Hybrid Flagellin Design Präsentiert vom slowenischen Team auf der iGEM 2008Überraschenderweise sind wir dank der neuesten Entwicklungen in der Genomik, der synthetischen Biologie und zuletzt in Cloud-Labors sehr nahe daran, jedes gewünschte Protein zu erstellen, ohne Jupyters Notizbuch zu verlassen.
In diesem Artikel werde ich Python-Code von der Idee eines Proteins bis zu seiner Expression in einer Bakterienzelle zeigen, ohne eine Pipette zu berühren oder mit jemandem zu sprechen. Die Gesamtkosten betragen nur ein paar hundert Dollar! Unter Verwendung der
Terminologie von Vijaya Pande aus A16Z ist dies Biologie 2.0.
Im folgenden Artikel führt der Python-Code des Cloud-Labors Folgendes aus:
- Synthese einer DNA-Sequenz, die jedes gewünschte Protein codiert.
- Klonierung dieser synthetischen DNA in einen Vektor , der sie exprimieren kann.
- Transformation von Bakterien mit diesem Vektor und Bestätigung, dass eine Expression stattfindet.
Python-Setup
Zunächst die allgemeinen Python-Einstellungen, die für jeden Jupyter-Notizblock erforderlich sind. Wir importieren einige nützliche Python-Module und erstellen einige Dienstprogrammfunktionen, hauptsächlich für die Datenvisualisierung.
Codeimport re import json import logging import requests import itertools import numpy as np import seaborn as sns import pandas as pd import matplotlib as mpl import matplotlib.pyplot as plt from io import StringIO from pprint import pprint from Bio.Seq import Seq from Bio.Alphabet import generic_dna from IPython.display import display, Image, HTML, SVG def uprint(astr): print(astr + "\n" + "-"*len(astr)) def show_html(astr): return display(HTML('{}'.format(astr))) def show_svg(astr, w=1000, h=1000): SVG_HEAD = '''<?xml version="1.0" standalone="no"?><!DOCTYPE svg PUBLIC "-//W3C//DTD SVG 1.1//EN" "http://www.w3.org/Graphics/SVG/1.1/DTD/svg11.dtd">''' SVG_START = '''<svg viewBox="0 0 {w:} {h:}" version="1.1" xmlns="http://www.w3.org/2000/svg" xmlns:xlink= "http://www.w3.org/1999/xlink">''' return display(SVG(SVG_HEAD + SVG_START.format(w=w, h=h) + astr + '</svg>')) def table_print(rows, header=True): html = ["<table>"] html_row = "</td><td>".join(k for k in rows[0]) html.append("<tr style='font-weight:{}'><td>{}</td></tr>".format('bold' if header is True else 'normal', html_row)) for row in rows[1:]: html_row = "</td><td>".join(row) html.append("<tr style='font-family:monospace;'><td>{:}</td></tr>".format(html_row)) html.append("</table>") show_html(''.join(html)) def clean_seq(dna): dna = re.sub("\s","",dna) assert all(nt in "ACGTN" for nt in dna) return Seq(dna, generic_dna) def clean_aas(aas): aas = re.sub("\s","",aas) assert all(aa in "ACDEFGHIKLMNPQRSTVWY*" for aa in aas) return aas def Images(images, header=None, width="100%"):
Cloud Labs
Wie AWS oder jede andere Computing-Cloud verfügt das Cloud-Labor über molekularbiologische Geräte sowie Roboter, die über das Internet geleast werden. Sie können Ihren Robotern Anweisungen erteilen, indem Sie auf einige Schaltflächen auf der Benutzeroberfläche klicken oder Code schreiben, der sie selbst programmiert. Es ist nicht notwendig, eigene Protokolle zu schreiben, wie ich es hier tun werde. Ein wesentlicher Teil der Molekularbiologie sind Standard-Routineaufgaben. Daher ist es normalerweise besser, sich auf ein zuverlässiges Alien-Protokoll zu verlassen, das eine gute Interaktion mit Robotern zeigt.
Vor kurzem sind eine Reihe von Unternehmen mit Cloud-Labors erschienen:
Transcriptic ,
Autodesk Wet Lab Accelerator (Beta und auf Basis von Transcriptic),
Arcturus BioCloud (Beta),
Emerald Cloud Lab (Beta),
Synthego (noch nicht gestartet). Es gibt sogar Unternehmen, die auf Cloud-Labors aufbauen, wie beispielsweise
Desktop Genetics , das auf CRISPR spezialisiert ist.
Wissenschaftliche Artikel über die Verwendung von Cloud Labs in der realen Wissenschaft beginnen zu erscheinen.
Zum Zeitpunkt dieses Schreibens ist nur Transcriptic gemeinfrei, daher werden wir es verwenden. Soweit ich weiß, basiert der größte Teil des Transcriptic-Geschäfts auf der Automatisierung gängiger Protokolle, und das Schreiben eigener Protokolle in Python (wie in diesem Artikel beschrieben) ist weniger verbreitet.
 Transkriptionelle „Arbeitszelle“ mit Kühlschränken unten und verschiedenen Laborgeräten am Stand
Transkriptionelle „Arbeitszelle“ mit Kühlschränken unten und verschiedenen Laborgeräten am StandIch werde Transcriptic Robots Anweisungen zum
Auto-Protokoll geben . Autoprotokoll ist eine JSON-basierte Sprache zum Schreiben von Protokollen für Laborroboter (und sozusagen für Menschen). Autoprotokoll wird hauptsächlich in
dieser Python-Bibliothek erstellt . Die Sprache wurde ursprünglich erstellt und wird immer noch von Transcriptic unterstützt, ist aber meines Wissens vollständig offen. Es gibt eine gute
Dokumentation .
Eine interessante Idee ist, dass Sie Anweisungen für Personen in entfernten Labors, beispielsweise in China oder Indien, über das Autoprotokoll schreiben können und möglicherweise einige Vorteile aus der Verwendung von Personen (deren Urteilsvermögen) und Robotern (mangelndes Urteilsvermögen) ziehen können. Wir müssen
hier protocols.io erwähnen. Dies ist ein Versuch, Protokolle zu standardisieren, um die Reproduzierbarkeit zu verbessern, aber für Menschen, nicht für Roboter.
"instructions": [ { "to": [ { "well": "water/0", "volume": "500.0:microliter" } ], "op": "provision", "resource_id": "rs17gmh5wafm5p" }, ... ]
Beispiel für ein AutoprotokollfragmentPython-Einstellungen für die Molekularbiologie
Zusätzlich zum Importieren von Standardbibliotheken benötige ich einige spezifische molekularbiologische Dienstprogramme. Dieser Code ist hauptsächlich für Autoprotokoll und Transkription vorgesehen.
Das Konzept des "Totvolumens" findet sich häufig im Code. Dies bedeutet den letzten Tropfen Flüssigkeit, den Transkriptionsroboter nicht mit einer Pipette aus den Röhrchen nehmen können (weil sie ihn nicht sehen können!). Sie müssen viel Zeit aufwenden, um sicherzustellen, dass die Flaschen genügend Material haben.
Code import autoprotocol from autoprotocol import Unit from autoprotocol.container import Container from autoprotocol.protocol import Protocol from autoprotocol.protocol import Ref
DNA-Synthese und synthetische Biologie
Trotz ihrer Verbindung mit der modernen synthetischen Biologie ist die DNA-Synthese eine ziemlich alte Technologie. Seit Jahrzehnten können wir Oligonukleotide herstellen (dh DNA-Sequenzen bis zu 200 Basen). Es war jedoch immer teuer und die Chemie erlaubte nie lange DNA-Sequenzen. In letzter Zeit ist es zu einem vernünftigen Preis möglich geworden, ganze Gene (bis zu Tausenden von Basen) zu synthetisieren. Diese Leistung eröffnet wirklich die Ära der „synthetischen Biologie“.
Die
synthetische Genomik von Craig Venter
hat die synthetische Biologie am weitesten vorangetrieben, indem sie
einen ganzen Organismus synthetisiert hat - mehr als eine Million Basen lang. Mit zunehmender Länge der DNA besteht das Problem nicht mehr in der Synthese, sondern in der Assemblierung (d. H. Zusammenfügen synthetisierter DNA-Sequenzen). Mit jeder Baugruppe können Sie die DNA-Länge (oder mehr) verdoppeln, sodass Sie nach etwa einem Dutzend Iterationen ein
ziemlich langes Molekül erhalten ! Die Unterscheidung zwischen Synthese und Zusammenbau sollte dem Endbenutzer bald klar werden.
Moores Gesetz?
Der Preis für die DNA-Synthese sinkt ziemlich schnell von über 0,30 USD vor zwei Jahren auf heute etwa 0,10 USD, aber sie entwickelt sich eher wie Bakterien als wie Prozessoren. Im Gegensatz dazu fallen die Preise für DNA-Sequenzierung schneller als das Gesetz von Moore. Ein Ziel von
0,02 USD pro Base wird als Wendepunkt festgelegt, an dem Sie viele zeitaufwändige DNA-Manipulationen durch einfache Synthese ersetzen können. Zu diesem Preis können Sie beispielsweise ein ganzes 3-kb-Plasmid für
60 US-Dollar synthetisieren und eine Menge Molekularbiologie überspringen. Ich hoffe, wir werden dies in ein paar Jahren erreichen.
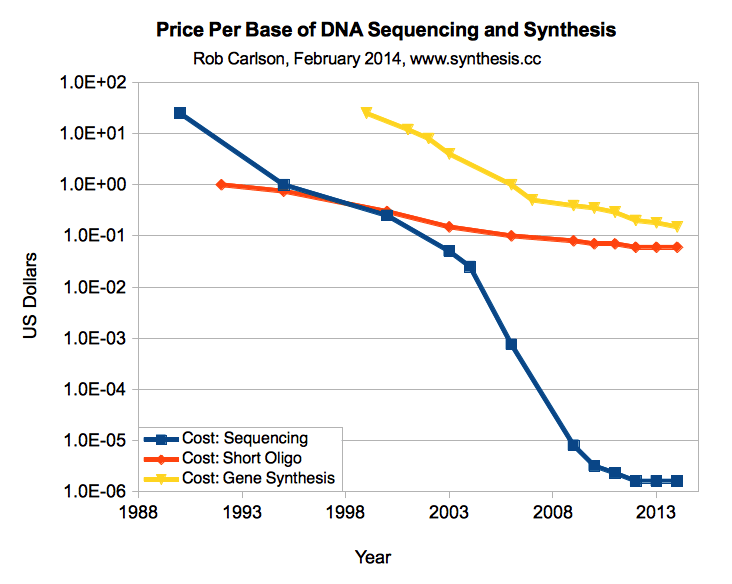 DNA-Synthesepreise im Vergleich zu DNA-Sequenzierungspreisen, Preis für 1 Base (Carlson, 2014)
DNA-Synthesepreise im Vergleich zu DNA-Sequenzierungspreisen, Preis für 1 Base (Carlson, 2014)DNA-Syntheseunternehmen
Auf dem Gebiet der DNA-Synthese gibt es mehrere große Unternehmen: IDT ist der größte Hersteller von Oligonukleotiden und kann auch längere (bis zu 2 kb) „Genfragmente“ (
gBlocks ) produzieren.
Gen9 ,
Twist und
DNA 2.0 sind normalerweise auf längere DNA-Sequenzen spezialisiert - dies sind Gensyntheseunternehmen. Es gibt auch einige interessante neue Unternehmen wie
Cambrian Genomics und
Genesis DNA , die an Synthesemethoden der nächsten Generation arbeiten.
Andere Unternehmen wie
Amyris ,
Zymergen und
Ginkgo Bioworks verwenden die von diesen Unternehmen synthetisierte DNA, um auf
Körperebene zu arbeiten.
Synthetic Genomics macht das auch, aber es synthetisiert die DNA selbst.
Ginkgo hat kürzlich
einen Deal mit Twist abgeschlossen , um 100 Millionen Basen zu machen: den größten Deal, den ich je gesehen habe. Dies beweist, dass wir in der Zukunft leben. Twist hat sogar einen Werbecode auf Twitter beworben: Wenn Sie 10 Millionen DNA-Basen (fast das gesamte Hefegenom!) Kaufen, erhalten Sie weitere 10 Millionen kostenlos.
 Twitter Twist Nischenangebot
Twitter Twist NischenangebotErster Teil: Versuchsaufbau
Grün fluoreszierendes Protein
In diesem Experiment synthetisieren wir eine DNA-Sequenz für ein einfaches,
grün fluoreszierendes Protein (GFP). Das GFP-Protein wurde zuerst in einer
Qualle gefunden , die unter ultraviolettem Licht fluoresziert. Dies ist ein äußerst nützliches Protein, da es leicht durch einfache Messung der Fluoreszenz nachgewiesen werden kann. Es gibt GFP-Optionen, die Gelb, Rot, Orange und andere Farben erzeugen.
Es ist interessant zu sehen, wie verschiedene Mutationen die Farbe eines Proteins beeinflussen, und dies ist ein potenziell interessantes Problem des maschinellen Lernens. In jüngerer Zeit mussten Sie dafür viel Zeit im Labor verbringen, aber jetzt werde ich Ihnen zeigen, dass es (fast) so einfach ist wie das Bearbeiten einer Textdatei!
Technisch gesehen ist mein GFP eine Super Folder Option (sfGFP) mit einigen Mutationen zur Verbesserung der Qualität.
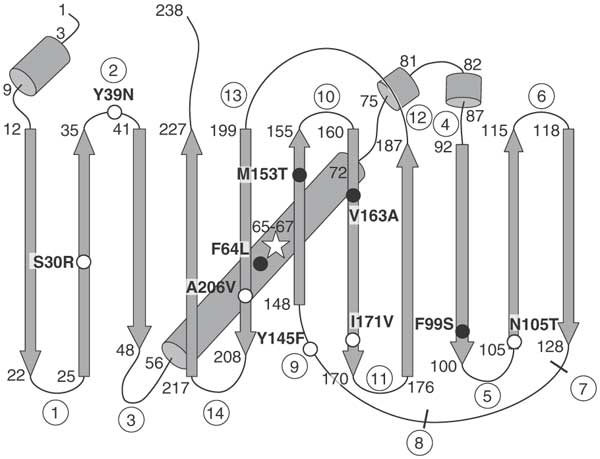 In Superfolder-GFP (sfGFP) geben einige Mutationen ihm bestimmte nützliche Eigenschaften.
In Superfolder-GFP (sfGFP) geben einige Mutationen ihm bestimmte nützliche Eigenschaften.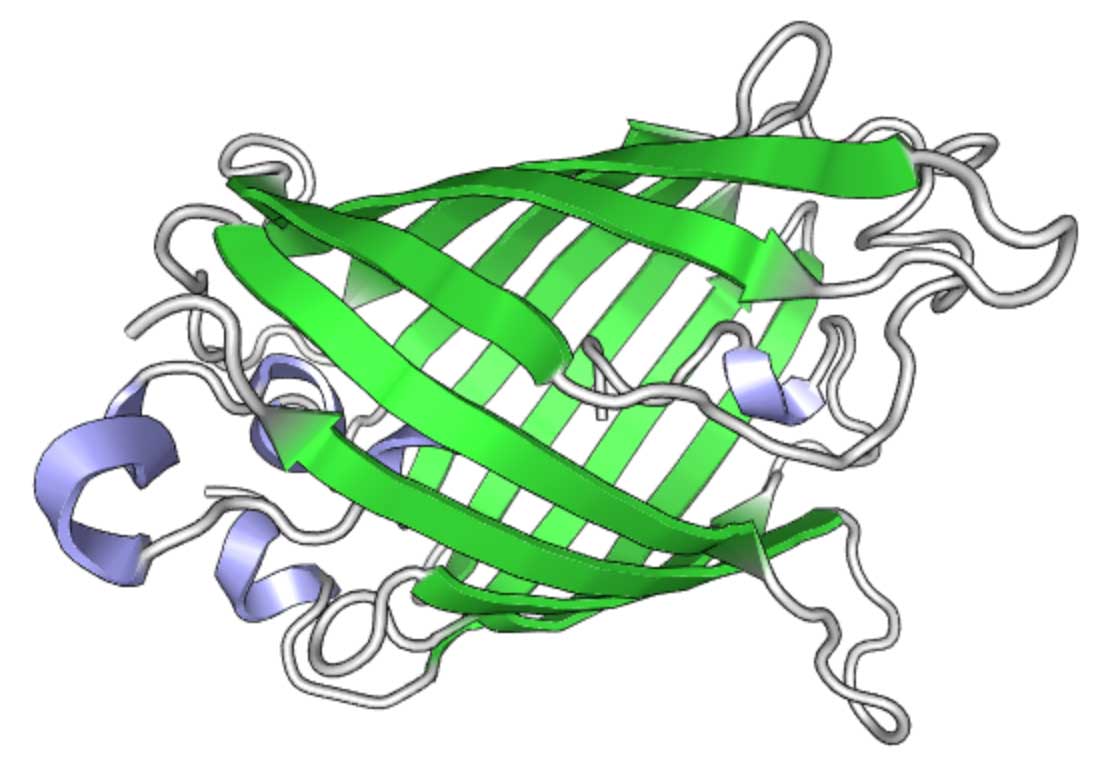 GFP-Struktur (visualisiert mit PV )
GFP-Struktur (visualisiert mit PV )GFP-Synthese in Twist
Ich hatte das Glück, in Twists Alpha-Testprogramm einzusteigen, also nutzte ich ihren DNA-Synthesedienst (sie gaben freundlicherweise meine winzige Bestellung auf - danke, Twist!). Dies ist ein neues Unternehmen auf unserem Gebiet mit einem neuen vereinfachten Syntheseverfahren. Ihre Preise liegen
bei 0,10 USD pro Basis oder darunter , aber sie befinden sich
noch in der Beta und das Alpha-Programm, an dem ich teilgenommen habe, wurde geschlossen. Twist hat ungefähr 150 Millionen US-Dollar gesammelt, daher ist ihre Technologie lebendig.
Ich habe meine DNA-Sequenz als Excel-Tabelle an Twist gesendet (es gibt noch keine API, aber ich denke, es wird bald soweit sein), und sie haben die synthetisierte DNA direkt an meine Box im Transkriptionslabor gesendet (ich habe auch IDT für die Synthese verwendet, aber sie haben nicht gesendet DNA direkt in Transcriptic, was den Spaß ein bisschen verdirbt).
Offensichtlich ist dieser Prozess noch kein typischer Anwendungsfall geworden und erfordert Unterstützung, aber er hat funktioniert, sodass die gesamte Pipeline virtuell bleibt. Ohne dies würde ich wahrscheinlich Zugang zum Labor benötigen - viele Unternehmen werden keine DNA oder Reagenzien an ihre Heimatadresse senden.
 GFP ist harmlos, daher wird jede Art hervorgehoben
GFP ist harmlos, daher wird jede Art hervorgehobenPlasmidvektor
Um dieses Protein in Bakterien zu exprimieren, muss das Gen irgendwo leben, sonst wird die synthetische DNA, die das Gen codiert, einfach sofort abgebaut. In der Molekularbiologie verwenden wir in der Regel ein Plasmid, ein Stück runder DNA, das außerhalb des Bakteriengenoms lebt und Proteine exprimiert. Plasmide sind eine bequeme Möglichkeit für Bakterien, nützliche, eigenständige Funktionsmodule wie Antibiotikaresistenz gemeinsam zu nutzen. In einer Zelle können sich Hunderte von Plasmiden befinden.
Die weit verbreitete Terminologie ist, dass ein Plasmid ein
Vektor ist und synthetische DNA eine Insertion (Insertion) ist. Hier versuchen wir also, die Insertion in einen Vektor zu klonen und dann die Bakterien unter Verwendung des Vektors zu transformieren.
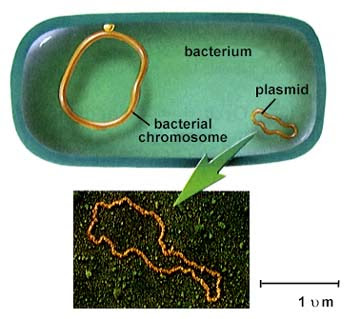 Bakteriengenom und Plasmid (nicht maßstabsgetreu!) ( Wikipedia )
Bakteriengenom und Plasmid (nicht maßstabsgetreu!) ( Wikipedia )pUC19
Ich habe ein ziemlich normales Plasmid in der
pUC19- Serie gewählt. Dieses Plasmid wird sehr oft verwendet, und da es als Teil des Standard-Transkriptinventars verfügbar ist, müssen wir ihnen nichts senden.
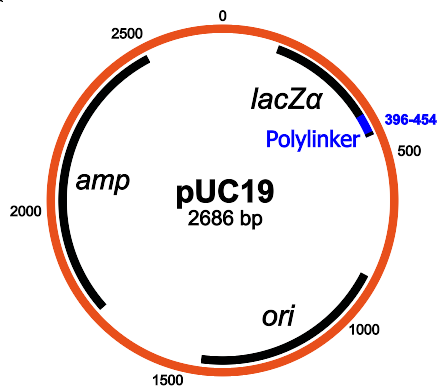 Struktur von pUC19: Die Hauptkomponenten sind das Ampicillin-Resistenzgen lacZα, MCS / Polylinker und der Replikationsursprung (Wikipedia).
Struktur von pUC19: Die Hauptkomponenten sind das Ampicillin-Resistenzgen lacZα, MCS / Polylinker und der Replikationsursprung (Wikipedia).PUC19 hat eine nette Funktion: Da es das lacZα-Gen enthält, können Sie die
blau-weiße Selektionsmethode verwenden und sehen, in welchen Kolonien die Insertion erfolgreich war. Es werden zwei Chemikalien benötigt:
IPTG und
X-Gal , und die Schaltung funktioniert wie folgt:
- IPTG induziert die lacZα-Expression.
- Wenn lacZα über DNA deaktiviert wird, die an der Mehrfachklonierungsstelle ( MCS / Polylinker ) in lacZα inseriert ist, kann das Plasmid X-Gal nicht hydrolysieren und diese Kolonien sind weiß statt blau.
- Daher erzeugt eine erfolgreiche Insertion weiße Kolonien und eine fehlgeschlagene Insertion blaue Kolonien.
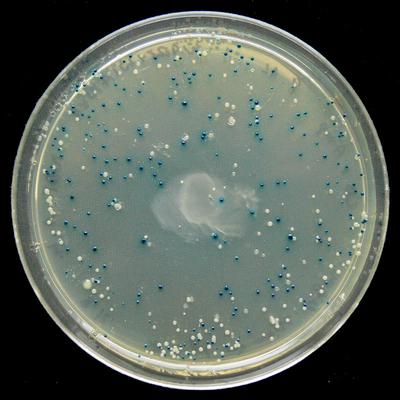 Die blau-weiße Auswahl zeigt, wo die lacZα-Expression deaktiviert wurde ( Wikipedia )In der OpenWetware-Dokumentation
Die blau-weiße Auswahl zeigt, wo die lacZα-Expression deaktiviert wurde ( Wikipedia )In der OpenWetware-Dokumentation heißt es:
E. coli DH5α benötigt kein IPTG, um die Expression des lac-Promotors zu induzieren, selbst wenn ein Lac-Repressor im Stamm exprimiert wird. Die Kopienzahl der meisten Plasmide übersteigt die Anzahl der Repressoren in den Zellen. Wenn Sie eine maximale Expression benötigen, fügen Sie IPTG bis zu einer Endkonzentration von 1 mM hinzu.
Synthetische DNA-Sequenzen
SfGFP-DNA-Sequenz
Es ist einfach, die DNA-Sequenz für sfGFP zu erhalten, indem
die Proteinsequenz genommen und
mit Codons codiert wird,
die für den Wirtsorganismus geeignet sind (hier
E. coli ). Dies ist ein mittelgroßes Protein mit 236 Aminosäuren, sodass die DNA-Synthese bei 10 Cent etwa
70 US-Dollar pro Base kostet.
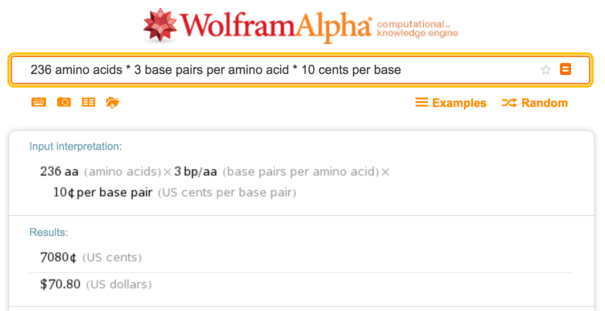 Wolfram Alpha, Synthesekostenberechnung
Wolfram Alpha, SynthesekostenberechnungDie ersten 12 Basen unseres sfGFP sind die
Shine-Delgarno-Sequenz , die ich selbst hinzugefügt habe und die theoretisch die Expression erhöhen sollte (AGGAGGACAGCT, dann startet ATG (
Startcodon ) das Protein). Nach einem von
Salis Lab entwickelten Rechenwerkzeug (
Vorlesungsfolien ) können wir eine mittlere bis hohe Expression unseres Proteins erwarten (Translationsinitiationsrate von 10.000 „willkürlichen Einheiten“).
sfGFP_plus_SD = clean_seq(""" AGGAGGACAGCTATGTCGAAAGGAGAAGAACTGTTTACCGGTGTGGTTCCGATTCTGGTAGAACTGGA TGGGGACGTGAACGGCCATAAATTTAGCGTCCGTGGTGAGGGTGAAGGGGATGCCACAAATGGCAAAC TTACCCTTAAATTCATTTGCACTACCGGCAAGCTGCCGGTCCCTTGGCCGACCTTGGTCACCACACTG ACGTACGGGGTTCAGTGTTTTTCGCGTTATCCAGATCACATGAAACGCCATGACTTCTTCAAAAGCGC CATGCCCGAGGGCTATGTGCAGGAACGTACGATTAGCTTTAAAGATGACGGGACCTACAAAACCCGGG CAGAAGTGAAATTCGAGGGTGATACCCTGGTTAATCGCATTGAACTGAAGGGTATTGATTTCAAGGAA GATGGTAACATTCTCGGTCACAAATTAGAATACAACTTTAACAGTCATAACGTTTATATCACCGCCGA CAAACAGAAAAACGGTATCAAGGCGAATTTCAAAATCCGGCACAACGTGGAGGACGGGAGTGTACAAC TGGCCGACCATTACCAGCAGAACACACCGATCGGCGACGGCCCGGTGCTGCTCCCGGATAATCACTAT TTAAGCACCCAGTCAGTGCTGAGCAAAGATCCGAACGAAAAACGTGACCATATGGTGCTGCTGGAGTT CGTGACCGCCGCGGGCATTACCCATGGAATGGATGAACTGTATAAA""") print("Read in sfGFP plus Shine-Dalgarno: {} bases long".format(len(sfGFP_plus_SD))) sfGFP_aas = clean_aas("""MSKGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLTYG VQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNFNSHNVYITADKQKN GIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHGMDELYK""") assert sfGFP_plus_SD[12:].translate() == sfGFP_aas print("Translation matches protein with accession 532528641")
Lesen Sie in sfGFP plus Shine-Dalgarno: 726 Basen lang
Die Übersetzung stimmt mit dem Protein 532528641 überein
PUC19-DNA-Sequenz
Zuerst überprüfe ich, ob die
von der NEB heruntergeladene pUC19-Sequenz die richtige Länge hat und den erwarteten
Polylinker enthält .
pUC19_fasta = !cat puc19fsa.txt pUC19_fwd = clean_seq(''.join(pUC19_fasta[1:])) pUC19_rev = pUC19_fwd.reverse_complement() assert all(nt in "ACGT" for nt in pUC19_fwd) assert len(pUC19_fwd) == 2686 pUC19_MCS = clean_seq("GAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTT") print("Read in pUC19: {} bases long".format(len(pUC19_fwd))) assert pUC19_MCS in pUC19_fwd print("Found MCS/polylinker")
Lesen Sie in pUC19: 2686 Basen lang
Gefunden MCS / Polylinker
Wir führen einige grundlegende QCs durch, um sicherzustellen, dass EcoRI und BamHI nur einmal in pUC19 vorhanden sind (die folgenden Restriktionsenzyme sind im Standard-Transkriptionsinventar verfügbar:
PstI ,
PvuII ,
EcoRI ,
BamHI ,
BbsI ,
BsmBI ).
REs = {"EcoRI":"GAATTC", "BamHI":"GGATTC"} for rename, res in REs.items(): assert (pUC19_fwd.find(res) == pUC19_fwd.rfind(res) and pUC19_rev.find(res) == pUC19_rev.rfind(res)) assert (pUC19_fwd.find(res) == -1 or pUC19_rev.find(res) == -1 or pUC19_fwd.find(res) == len(pUC19_fwd) - pUC19_rev.find(res) - len(res)) print("Asserted restriction enzyme sites present only once: {}".format(REs.keys()))
Jetzt schauen wir uns die lacZα-Sequenz an und stellen sicher, dass nichts Unerwartetes vorliegt. Zum Beispiel sollte es mit Met beginnen und mit einem Stoppcodon enden. Es ist auch leicht zu bestätigen, dass dies der vollständige 324 bp lacZα ORF ist, indem die pUC19-Sequenz in den freien
Snapgen-Viewer geladen wird .
lacZ = pUC19_rev[2217:2541] print("lacZα sequence:\t{}".format(lacZ)) print("r_MCS sequence:\t{}".format(pUC19_MCS.reverse_complement())) lacZ_p = lacZ.translate() assert lacZ_p[0] == "M" and not "*" in lacZ_p[:-1] and lacZ_p[-1] == "*" assert pUC19_MCS.reverse_complement() in lacZ assert pUC19_MCS.reverse_complement() == pUC19_rev[2234:2291] print("Found MCS once in lacZ sequence")
lacZ-Sequenz: ATGACCATGATTACGCCAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGTAATAGCGAAGAGGCCCGCACCGATCGCCCTTCCCAACAGTTGCGCAGCCTGAATGGCGAATGGCGCCTGATGCGGTATTTTCTCCTTACGCATCTGTGCGGTATTTCACACCGCATATGGTGCACTCTCAGTACAATCTGCTCTGATGCCGCATAG
r_MCS-Sequenz: AAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTC
MCS einmal in lacZ-Sequenz gefunden
Gibson Montage
DNA zusammenzusetzen bedeutet einfach, Fragmente zu vernetzen. Normalerweise sammeln Sie mehrere DNA-Fragmente in einem längeren Segment und klonieren es dann in ein Plasmid oder Genom. In diesem Experiment möchte ich ein DNA-Segment in das pUC19-Plasmid unterhalb des lac-Promotors zur Expression in
E. coli klonieren.
Es gibt viele Klonierungsmethoden (z. B.
NEB ,
OpenWetware ,
Addgen ). Hier werde ich die Gibson-Baugruppe verwenden (
die 2009 von Daniel Gibson in Synthetic Genomics entwickelt wurde), die nicht unbedingt die billigste Methode ist, aber einfach und flexibel. Sie müssen nur die DNA, die Sie sammeln möchten (mit der entsprechenden Überlappung), in ein Reagenzglas mit dem Gibson Assembly Master Mix geben, und es wird sich selbst zusammensetzen!
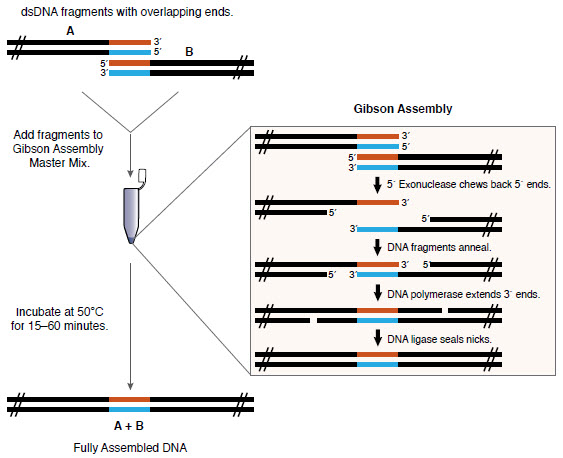 Gibson Assembly Review ( NEB )
Gibson Assembly Review ( NEB )Ausgangsmaterial
Wir beginnen mit 100 ng synthetischer DNA in 10 μl Flüssigkeit. Dies entspricht 0,21 Picomol DNA oder einer Konzentration von 10 ng / μl.
pmol_sfgfp = convert_ug_to_pmol(0.1, len(sfGFP_plus_SD)) print("Insert: 100ng of DNA of length {:4d} equals {:.2f} pmol".format(len(sfGFP_plus_SD), pmol_sfgfp))
Insert: 100 ng DNA mit einer Länge von 726 entsprechen 0,21 pmol
Nach
dem NEB-Montageprotokoll ist dies genug Quellmaterial:
Die NEB empfiehlt insgesamt 0,02 bis 0,5 Picomol DNA-Fragmente, wenn 1 oder 2 Fragmente zu dem Vektor zusammengesetzt werden, oder 0,2 bis 1,0 Picomol DNA-Fragmente, wenn 4 bis 6 Fragmente gesammelt werden.
0,02-0,5 pmol * X & mgr; l
* Die optimierte Klonierungseffizienz beträgt 50-100 ng Vektoren mit einem 2-3-fachen Überschuss an Insertionen. Verwenden Sie fünfmal mehr Einfügungen, wenn die Größe weniger als 200 Bit / s beträgt. Das Gesamtvolumen an ungefilterten PCR-Fragmenten in der Gibson-Assemblierungsreaktion sollte 20% nicht überschreiten.
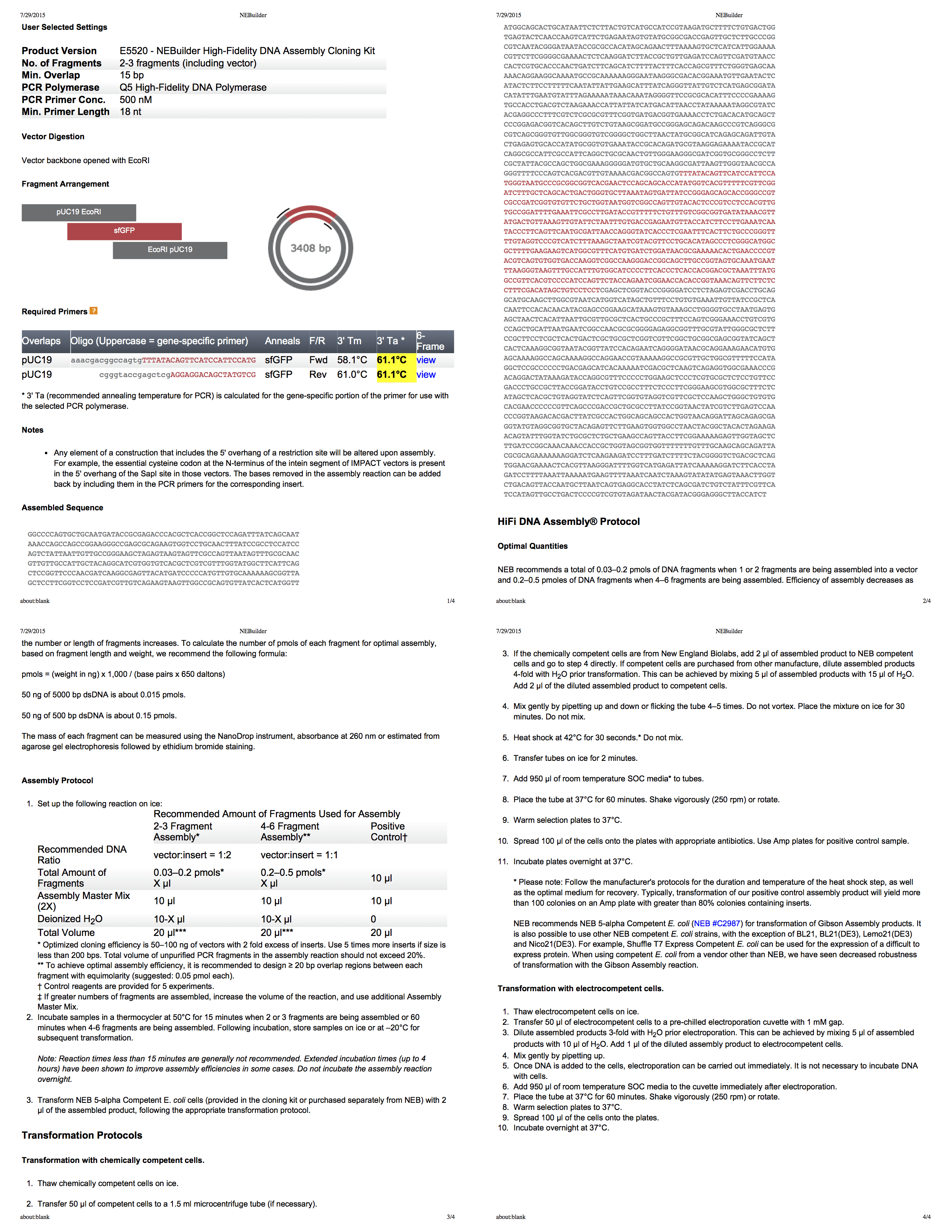
NEBuilder für Gibson-Montage
Der NEBuilder von Biolab ist ein wirklich großartiges Tool zum Erstellen des Gibson-Build-Protokolls. Sie erhalten sogar ein umfassendes vierseitiges PDF mit allen Informationen. Mit diesem Tool entwickeln wir ein Protokoll zum Schneiden von pUC19 mit EcoRI und verwenden dann PCR [PCR, Polymerasekettenreaktion ermöglicht einen signifikanten Anstieg kleiner Konzentrationen bestimmter DNA-Fragmente in biologischem Material - ca. per.], um der Insertion Fragmente der entsprechenden Größe hinzuzufügen.
Teil zwei: Experiment
Das Experiment besteht aus vier Phasen:
- Polymeraseketteninsertionsreaktion zur Zugabe von Material mit einer flankierenden Sequenz.
- Schneiden eines Plasmids zur Aufnahme.
- Assemblierung durch Gibson-Insertion und Plasmide.
- Transformation von Bakterien unter Verwendung des zusammengesetzten Plasmids.
Schritt 1. PCR-Insertion
Die Gibson-Assemblierung hängt von der DNA-Sequenz ab, die Sie sammeln, und weist eine überlappende Sequenz auf (siehe NEB-Protokoll mit detaillierten Anweisungen oben). Zusätzlich zur einfachen Amplifikation können Sie mit der PCR auch eine flankierende DNA-Sequenz hinzufügen, indem Sie einfach eine zusätzliche Sequenz in die Primer aufnehmen (kann auch
nur mit OE-PCR kloniert
werden ).
Wir synthetisieren Primer gemäß dem obigen NEB-Protokoll. Ich habe
das Schnellstartprotokoll auf der Transcriptic-Site ausprobiert, aber es gibt immer noch
einen Autoprotokollbefehl . Transcriptic selbst synthetisiert keine Oligonukleotide. Nach 1-2 Tagen Wartezeit erscheinen diese Primer auf magische Weise in meinem Inventar (beachten Sie, dass der genspezifische Teil der Primer unten in Großbuchstaben angegeben ist, dies sind jedoch nur kosmetische Dinge).
insert_primers = ["aaacgacggccagtgTTTATACAGTTCATCCATTCCATG", "cgggtaccgagctcgAGGAGGACAGCTATGTCG"]
Primer-Analyse
Sie können die Eigenschaften dieser Primer mit dem
IDT OligoAnalyzer analysieren . PCR
primer dimer , NEB .
Gene-specific portion of flank (uppercase)
Melt temperature: 51C, 53.5C
Full sequence
Melt temperature: 64.5C, 68.5C
Hairpin: -.4dG, -5dG
Self-dimer: -9dG, -16dG
Heterodimer: -6dG
PCR, , PCR. ( ), : . . , — .
Code """ PCR overlap extension of sfGFP according to NEB protocol. v5: Use 3/10ths as much primer as the v4 protocol. v6: more complex touchdown pcr procedure. The Q5 temperature was probably too hot v7: more time at low temperature to allow gene-specific part to anneal v8: correct dNTP concentration, real touchdown """ p = Protocol()
WARNING:root:Low volume for well sfGFP 1 /sfGFP 1 : 2.0:microliter
sfGFP 1 /sfGFP 1 2.0:microliter {'dilution': '0.25ng/ul'}
sfgfp_pcroe_v5_puc19_primer1_10uM 75.0:microliter {}
sfgfp_pcroe_v5_puc19_primer2_10uM 75.0:microliter {}
Consolidated volume 52.0:microliter
Protocol 1. Amplify the insert (oligos previously synthesized)
---------------------------------------------------------------
✓ Protocol analyzed
11 instructions
8 containers
Total Cost: $32.18
Workcell Time: $4.32
Reagents & Consumables: $27.86 : PCR
( )
( ). , , .
D1, E1, F1 2 , 4 8 . (50 ). , .
GelEval , , , , . . GelEval 40 /.
,
, dNTP , , 12,5 , 6 740bp 25 . GelEval 40 x 25 (1 2 ), , .
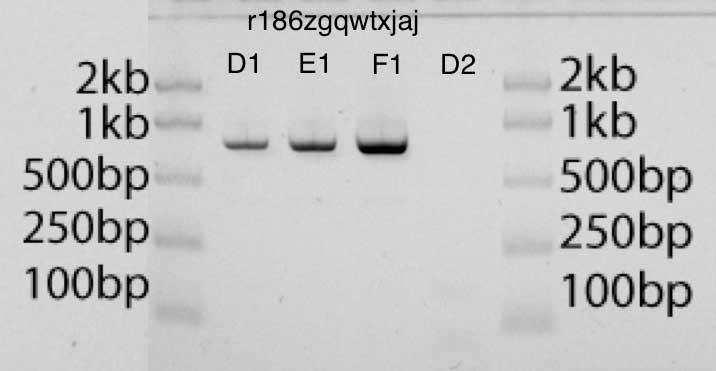 - EcoRI- pUC19, (D1, E1, F1), (D2)
- EcoRI- pUC19, (D1, E1, F1), (D2)PCR
Transcriptic . , .
, . 35 PCR,
PCR . — ! — , PCR , .
 PCR: , 35 42
PCR: , 35 422.
sfGFP pUC19, . NEB,
EcoRI . Transcriptic , :
NEB EcoRI 10x CutSmart ,
NEB pUC19 .
, . , Transcriptic :
Item ID Amount Concentration Price
------------ ------ ------------- ----------------- ------
CutSmart 10x B7204S 5 ml 10 X $19.00
EcoRI R3101L 50,000 units 20,000 units/ml $225.00
pUC19 N3041L 250 µg 1,000 µg/ml $268.00
NEB:
. 10X dH2O 1X. , , , , . 50 5 10x NEBuffer , dH2O.
, 1 λ 1 37°C 50 . , 5-10 10-20 1- .
1 50 .
Code """Protocol for cutting pUC19 with EcoRI.""" p = Protocol() experiment_name = "puc19_ecori_v3" options = {} inv = { 'water': "rs17gmh5wafm5p",
Volumes: re_tube:135.0:microliter water_tube:383.0:microliter EcoRI:30.0:microliter
Consolidated volume: 78.0:microliter
✓ Protocol analyzed
12 instructions
5 containers
Total Cost: $30.72
Workcell Time: $3.38
Reagents & Consumables: $27.34
:
, . .
«» ( 1,5 15 !). , D1 E1 ( ). , EcoRI .
, D1 E1 2,6kb. D2 : , .
-. , Transcriptic .
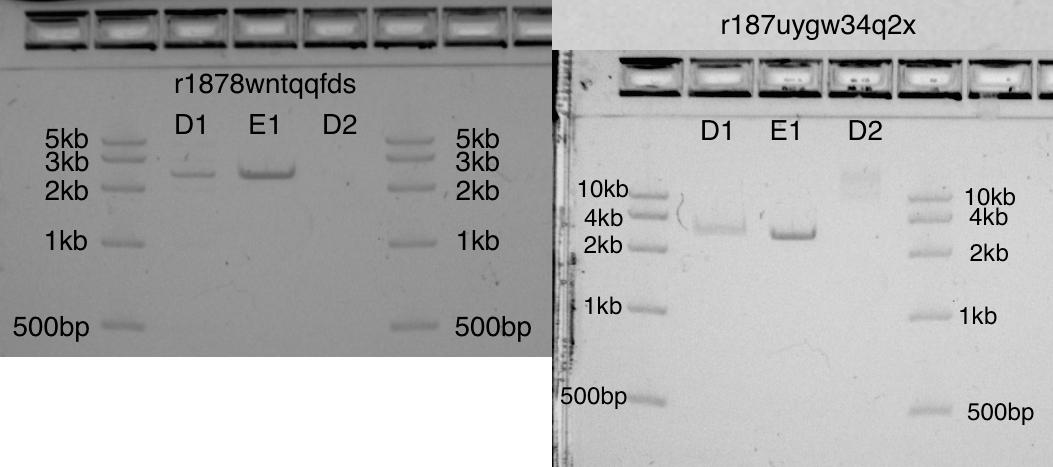 , pUC19 (2,6kb) D1 E1, pUC19 D2
, pUC19 (2,6kb) D1 E1, pUC19 D23.
, — ,
M13 ( )
qPCR , , . , , , .
, M13 , M13.
Code """Debugging transformation protocol: Gibson assembly followed by qPCR and a gel v2: include v3 Gibson assembly""" p = Protocol() options = {} experiment_name = "debug_sfgfp_puc19_gibson_seq_v2" inv = { "water" : "rs17gmh5wafm5p",
WARNING:root:Low volume for well sfgfp_puc19_gibson_v1_clone/sfgfp_puc19_gibson_v1_clone : 11.0:microliter
✓ Protocol analyzed
11 instructions
6 containers
Total Cost: $32.09
Workcell Time: $6.98
Reagents & Consumables: $25.11
: qPCR
qPCR JSON Transcriptic API.
, . API , .
-, :
project_id, run_id = "p16x6gna8f5e9", "r18mj3cz3fku7" api_url = "https://secure.transcriptic.com/hgbrian/{}/runs/{}/data.json".format(project_id, run_id) data_response = requests.get(api_url, headers=tsc_headers) data = data_response.json()
id, «» qPCR:
qpcr_id = data['debug_sfgfp_puc19_gibson_seq_v1_qpcr']['id'] pp_api_url = "https://secure.transcriptic.com/data/{}.json?key=postprocessed_data".format(qpcr_id) data_response = requests.get(pp_api_url, headers=tsc_headers) pp_data = data_response.json()
Ct ( ) . Ct — , . , (, , , ).
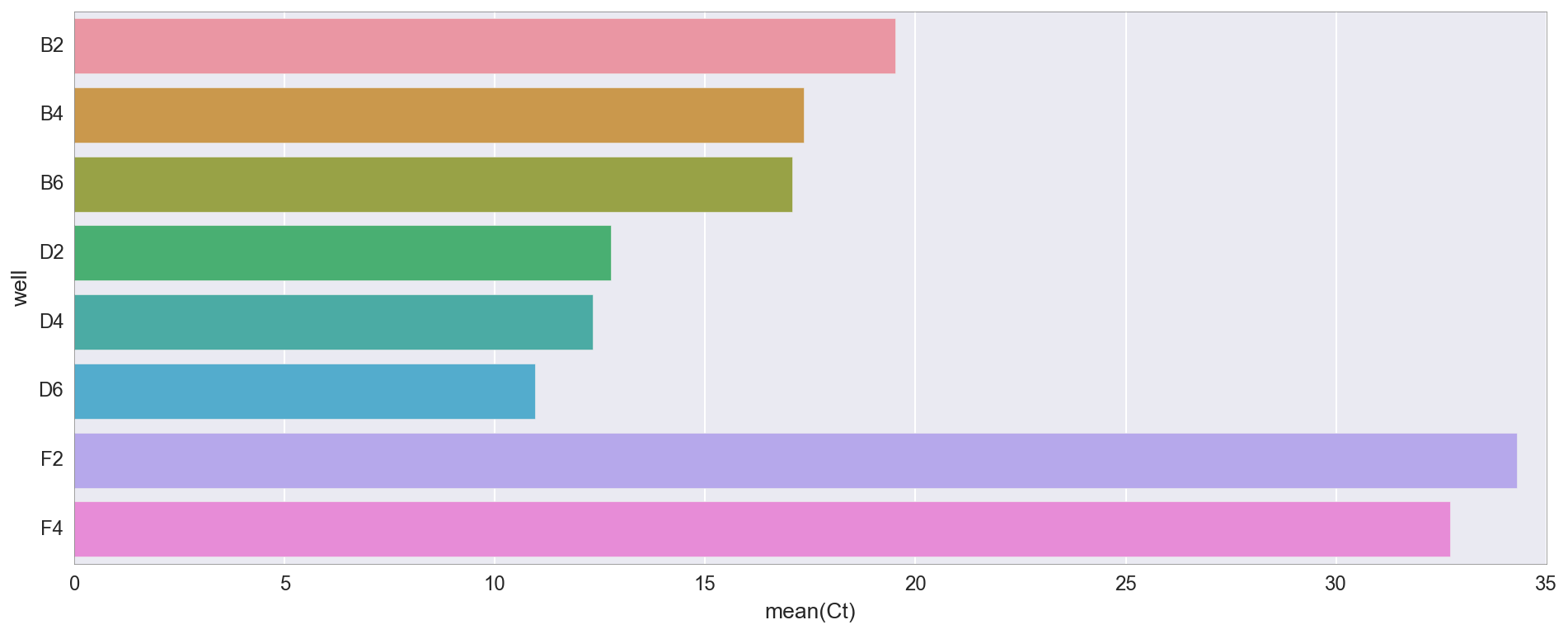
, D2/4/6 ( «v3»), B2/4/6 ( «v1»). v1 v3 , v3 4X NEB, . 30 (F2, F4), -, , .
qPCR, .
f, ax = plt.subplots(figsize=(16,6)) ax.set_color_cycle(['#fb6a4a', '#de2d26', '#a50f15', '#74c476', '#31a354', '#006d2c', '#08519c', '#6baed6']) amp0 = pp_data['amp0']['SYBR']['baseline_subtracted'] _ = [plt.plot(amp0[w_n[well]], label=well) for well in ['B2', 'B4', 'B6', 'D2', 'D4', 'D6', 'F2', 'F4']] _ = ax.set_ylim(0,) _ = plt.title("qPCR (reds=Gibson v1, greens=Gibson v3, blues=control)") _ = plt.legend(bbox_to_anchor=(1, .75), bbox_transform=plt.gcf().transFigure)
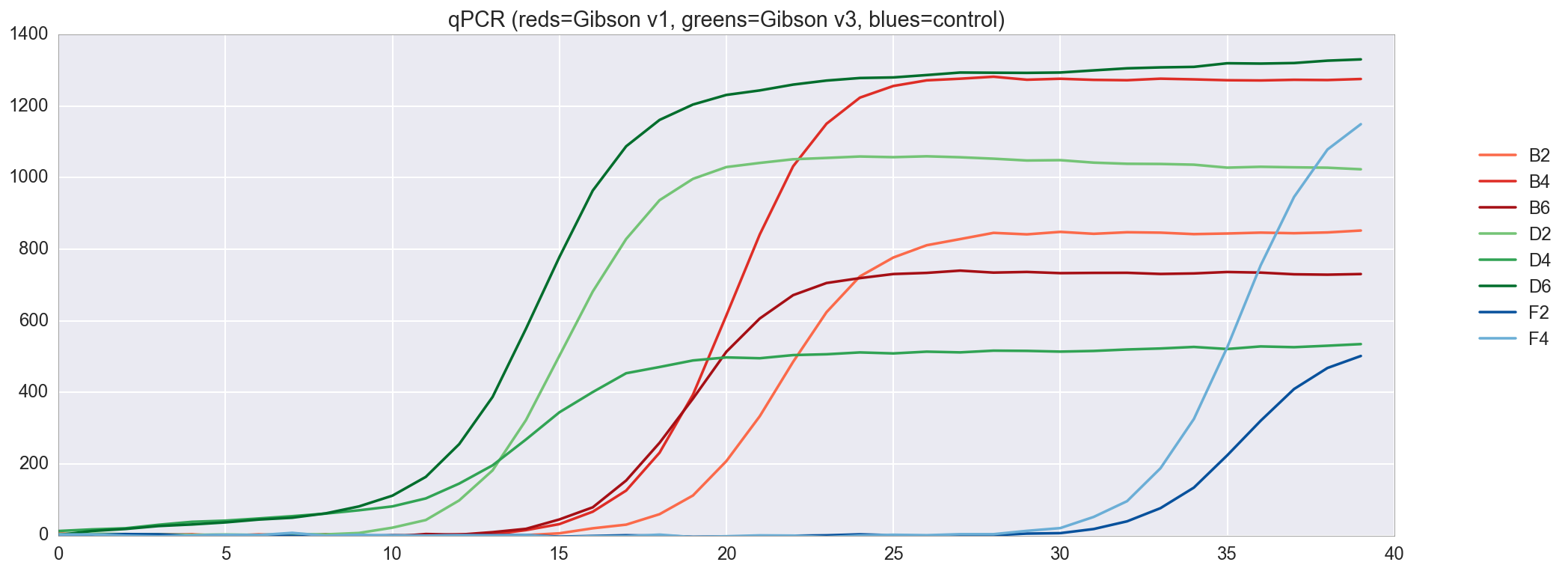
, qPCR , . v3 , v1, .
:
, 1kb B2, B4, B6, D2, D4, D6: ( 740bp, M13 — 40bp ). . , F2 F4 .
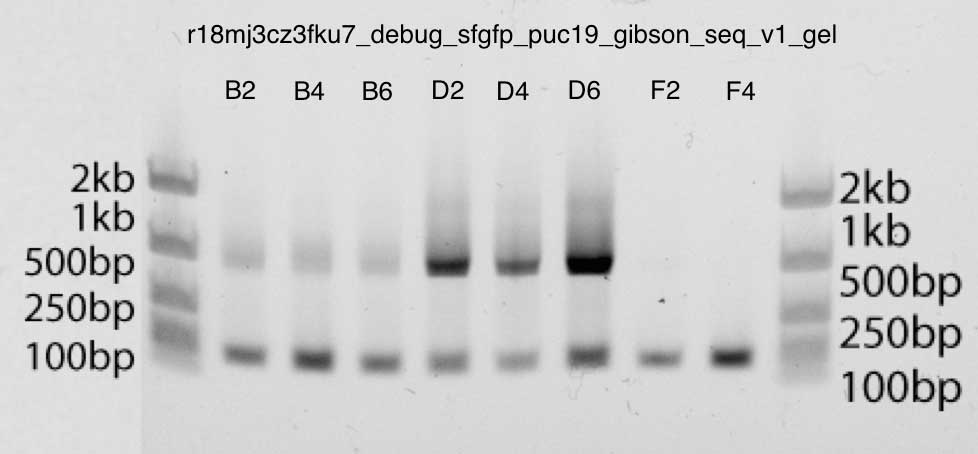 : v3 (D2, D4, D6), qPCR
: v3 (D2, D4, D6), qPCR4.
— .
E. coli sfGFP- pUC19.
Zymo DH5α Mix&Go . — Transcriptic. , , , , . , , .
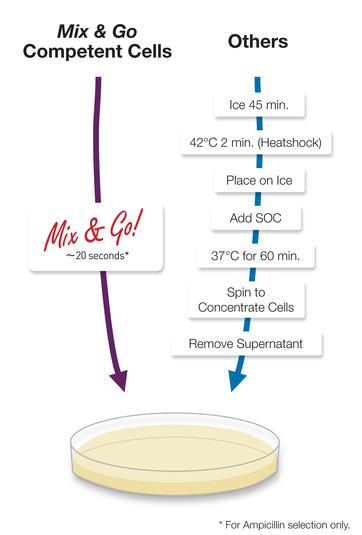 Zymo Mix & Go
Zymo Mix & Go— , . (« »), , («, »). , .
. , 37°C. , , , , Transcriptic — , . , , - , . . .
: (, , Mix&Go ); (, ); (, PCR ).
, , , , . , , !
, , , pUC19 (. . sfGFP) . pUC19 , , .
(«6-flat» Transcriptic), , . , , , . .
Code """Simple transformation protocol: transformation with unaltered pUC19""" p = Protocol() experiment_name = "debug_sfgfp_puc19_gibson_v1" inv = { "water" : "rs17gmh5wafm5p",
✓ Protocol analyzed
43 instructions
3 containers
$45.43
:
, ( ) , , . , Transcriptic , .
( ) , . , , , , 55 10 . . , . , , .
( , , ,
E. coli . , , ).
, , , .
 , pUC19, 18 : () ()
, pUC19, 18 : () ()pUC19, , , , sfGFP.
, IPTG X-gal,
- . , pUC19, sfGFP, .
, sfGFP 485 / 510 . , Transcriptic 485/535. , 485 510 . 600 (
OD600 ).
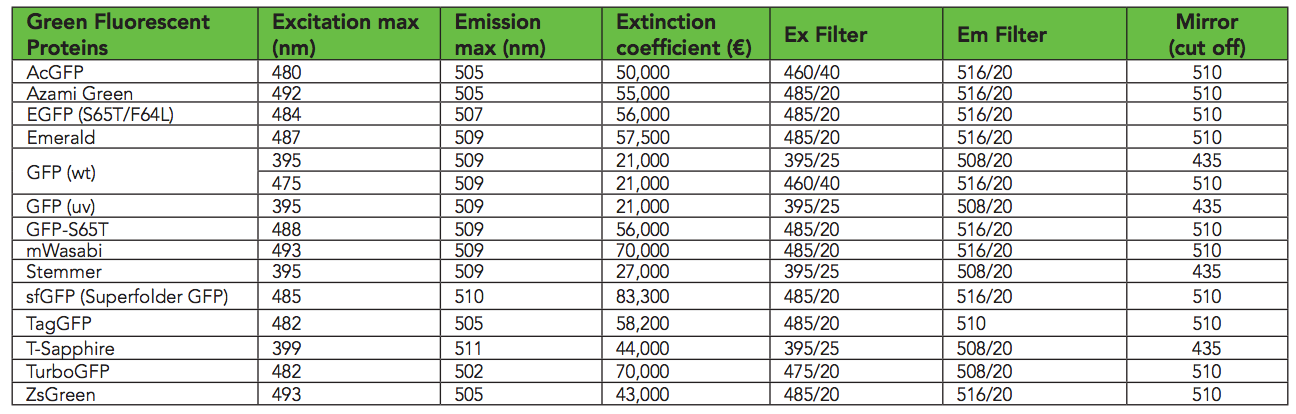 GFP ( biotek )
GFP ( biotek )IPTG X-gal
IPTG 1M 1:1000. , X-gal 20 / 1:1000 (20 /). , 2000µl LB 2 .
40 X-gal 20 / 40 IPTG 0,1 mM ( 4 IPTG 1M), 30 . , IPTG, X-gal .
Code """Full Gibson assembly and transformation protocol for sfGFP and pUC19 v1: Spread IPTG and X-gal onto plates, then spread cells v2: Mix IPTG, X-gal and cells; spread the mixture v3: exclude X-gal so I can do colony picking better v4: repeat v3 to try other excitation/emission wavelengths""" p = Protocol() options = { "gibson" : False,
Inventory: IPTG/IPTG/IPTG/IPTG/IPTG/IPTG 832.0:microliter {}
Inventory: sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone 57.0:microliter {}
✓ Protocol analyzed
40 instructions
8 containers
Total Cost: $53.20
Workcell Time: $17.35
Reagents & Consumables: $35.86 , «» 96- . (
autopick ).
Code """Pick colonies from plates and grow in amp media and check for fluorescence. v2: try again with a new plate (no blue colonies) v3: repeat with different emission and excitation wavelengths""" p = Protocol() options = {} for k, v in list(options.items()): if v is False: del options[k] experiment_name = "sfgfp_puc19_gibson_pick_v3" def plate_expid(val): """refer to the previous plating experiment's outputs""" plate_exp = "sfgfp_puc19_gibson_plates_v4" return "{}_{}".format(plate_exp, val)
✓ Protocol analyzed
62 instructions
8 containers
Total Cost: $66.38
Workcell Time: $57.59
Reagents & Consumables: $8.78
:
– , , (1-4) (5-6). , , , , IPTG X-gal, Transcriptic.
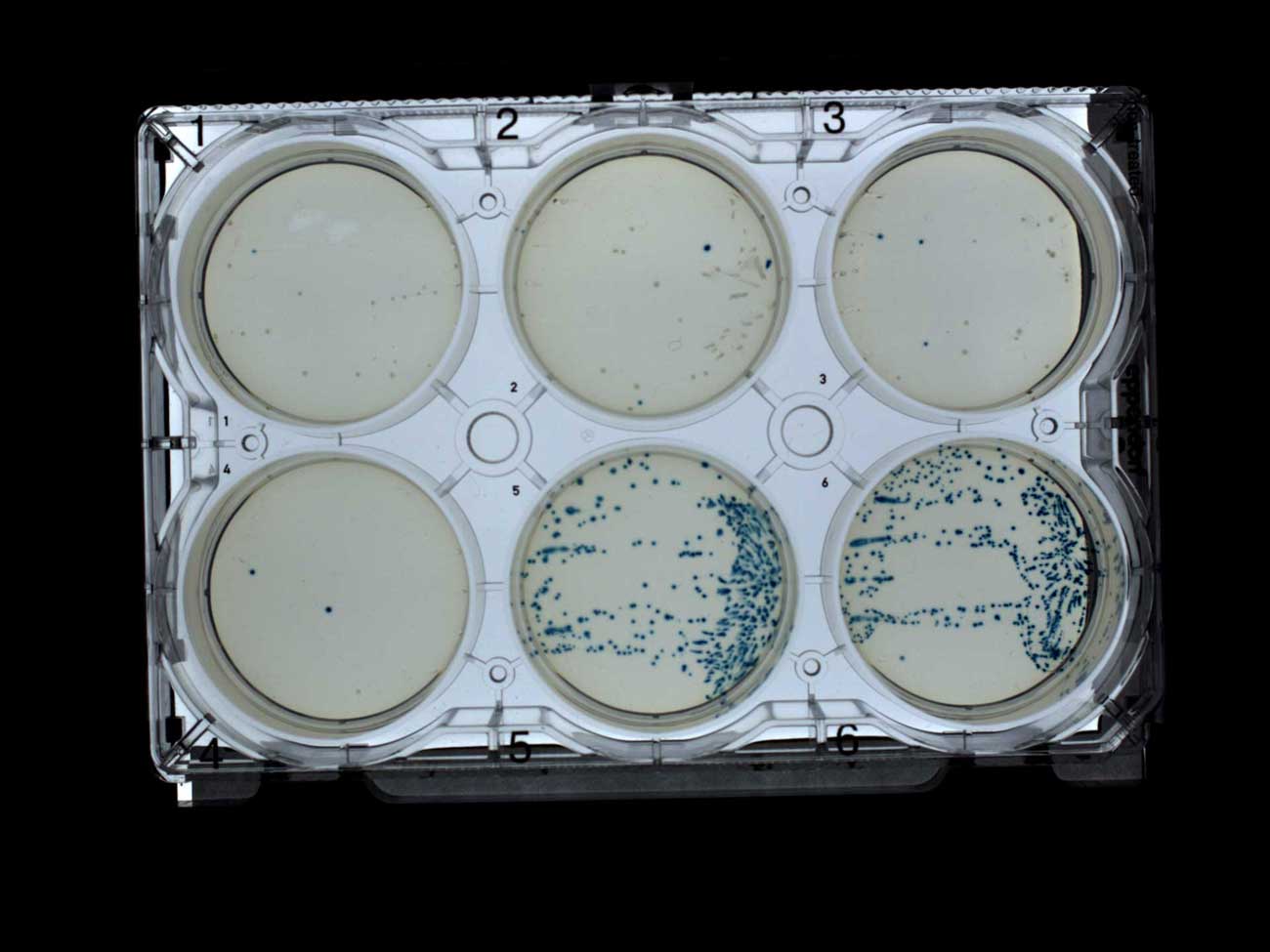 - (1-4) (5-6)
- (1-4) (5-6)- - . (
GraphicsMagick ). , , ( , ).
, Transcriptic. , 10 . , , . . , , , , .
 - (1-4) (5-6),
- (1-4) (5-6),– . , . , . X-gal.
- . , .
 , (1-4) (5-6)
, (1-4) (5-6):
50 96- 20 , sfGFP. ( , ) Transcriptic
Tecan Infinite .
, , , sfGFP. , , - , sfGFP . , sfGFP, , , , .
(OD600) 20 ( 60 ).
for t in [0,4,8,12,16,20]: abs_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_abs_{}.csv".format(t), index_col="Well") flr_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_fl2_{}.csv".format(t), index_col="Well") if t == 0: new_data = abs_data.join(flr_data) else: new_data = new_data.join(abs_data, rsuffix='_{}'.format(t)) new_data = new_data.join(flr_data, rsuffix='_{}'.format(t)) new_data.columns = ["OD 600:nanometer_0", "Fluorescence_0"] + list(new_data.columns[2:])
20- . , .
svg = [] W, H = 800, 500 min_x, max_x = 0, 0.8 min_y, max_y = 0, 50000 def _toxy(x, y): return W*(x-min_x)/(max_x-min_x), HH*(y-min_y)/(max_y-min_y) def _topt(x, y): return ','.join(map(str,_toxy(x,y))) ab_fls = [[row[0]] + [list(row[1])] for row in new_data.iterrows()]
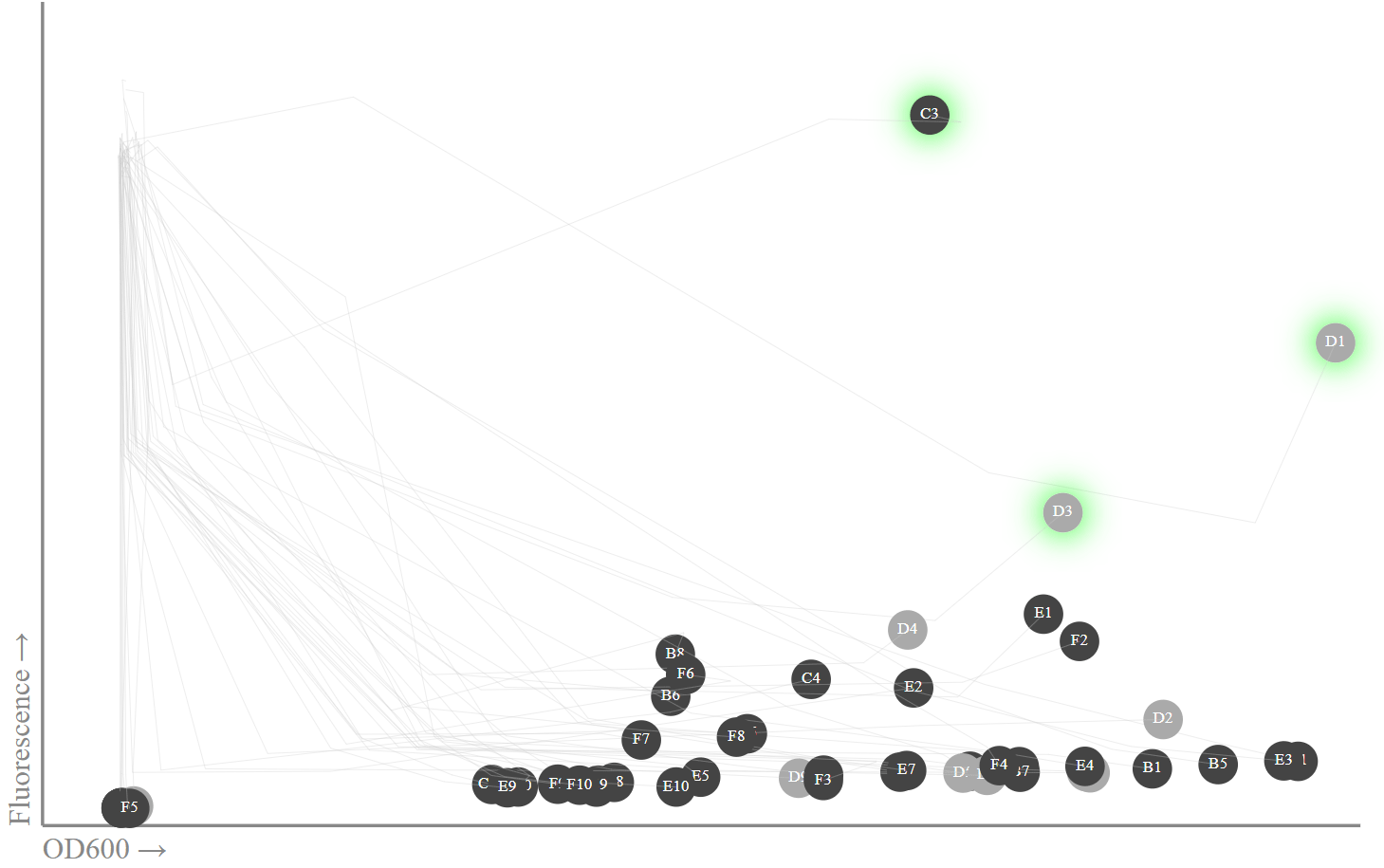 OD600: , . , sfGFPminiprep
OD600: , . , sfGFPminiprep , , 13. , - miniprep - Transcriptic, . (C1, D1, D3) (B1, B3, E1), sfGFP
muscle .
C1, D3 D3 sfGFP, B1, B3 E1 .
, . , 0 (40 000 ). 20- OD600 (, - ), . , , , , 11-15 .
(. . , ), , , ).
, , 50 sfGFP . , . , ( , , miniprep), 200 , .
, . , GFP, Python!
:
Preis
, , $360, :
- $70
- $32 PCR
- $31
- $32
- $53
- $67
- $75 3 miniprep'
, $250-300 . , 50 , , .
, ( ) ( IT). Transcriptic , . , , . , , , , .
, . , - , . , : , , IDT .
:
, . , :
- ! , . autoprotocol, .
- . 100 , .
- , , PCR. , , ? / ? , , , « 2-3 ». ?
- . . , .
- . .
- . . , 1 96 (96−x) 96- , .
- . csv , .
- . - , .
, , , . , 1994 :
- Transcriptic — . , , , . , , .
- — Transcriptic.
- , . Transcriptic ( , , ).
- , ( : ~$0). , .
- Transcriptic . , , .
, , - , , .
, :
- Twist/IDT/Gen9 Transcriptic (, - ).
- , , , , . .
- ( NEB, IDT) (, primer3 ).
(
) , ,
. , in vivo (. . ).
, , : RBS , ; ; .
?
, . :
- - //, .
- , , .
- in vivo split-GFP .
- scFv . scFvs - .
- BiTE , ( , ).
- , ( ).
- , . 1000 10 000 ? , GFP?
, ,
iGEM .
Transcriptic .