Wie man diesen Artikel liest : Ich entschuldige mich dafür, dass der Text so lang und chaotisch geworden ist. Um Ihnen Zeit zu sparen, beginne ich jedes Kapitel mit der Einführung von „Was ich gelernt habe“, in der ich die Essenz des Kapitels in ein oder zwei Sätzen erkläre.
"Zeigen Sie einfach die Lösung!" Wenn Sie nur sehen möchten, wozu ich gekommen bin, lesen Sie das Kapitel "Erfinderischer werden". Ich finde es jedoch interessanter und nützlicher, über Fehler zu lesen.Kürzlich wurde ich angewiesen, ein Verfahren zur Verarbeitung eines großen Volumens der ursprünglichen DNA-Sequenzen einzurichten (technisch gesehen ist dies ein SNP-Chip). Es war notwendig, schnell Daten über einen bestimmten genetischen Ort (SNP genannt) für die nachfolgende Modellierung und andere Aufgaben zu erhalten. Mit Hilfe von R und AWK konnte ich die Daten auf natürliche Weise bereinigen und organisieren und so die Bearbeitung von Anfragen erheblich beschleunigen. Dies war für mich nicht einfach und erforderte zahlreiche Iterationen. Dieser Artikel wird Ihnen helfen, einige meiner Fehler zu vermeiden und zu demonstrieren, was ich am Ende getan habe.
Zunächst einige einleitende Erklärungen.
Daten
Unser Genetic Information Processing Center der Universität hat uns 25 TB TSV-Daten zur Verfügung gestellt. Ich habe sie in 5 von Gzip komprimierte Pakete aufgeteilt, von denen jedes ungefähr 240 Vier-Gigabyte-Dateien enthielt. Jede Zeile enthielt Daten für einen SNP einer Person. Insgesamt wurden Daten zu ~ 2,5 Millionen SNPs und ~ 60.000 Menschen übertragen. Zusätzlich zu den SNP-Informationen gab es in den Dateien zahlreiche Spalten mit Zahlen, die verschiedene Merkmale wie Leseintensität, Häufigkeit verschiedener Allele usw. widerspiegeln. Es gab ungefähr 30 Spalten mit eindeutigen Werten.
Zweck
Wie bei jedem Datenverwaltungsprojekt war es am wichtigsten, zu bestimmen, wie die Daten verwendet werden sollen. In diesem Fall werden
wir zum größten Teil Modelle und Workflows für SNP basierend auf SNP auswählen . Das heißt, gleichzeitig benötigen wir Daten für nur einen SNP. Ich musste lernen, wie man alle Datensätze, die sich auf einen der 2,5 Millionen SNPs beziehen, so einfach wie möglich, schneller und billiger extrahiert.
Wie man es nicht macht
Ich werde ein passendes Klischee zitieren:
Ich habe nicht tausendmal versagt, sondern nur tausend Möglichkeiten entdeckt, eine Reihe von Daten nicht in einem für Abfragen geeigneten Format zu analysieren.
Erster Versuch
Was ich gelernt habe : Es gibt keine billige Möglichkeit, 25 TB gleichzeitig zu analysieren.
Nachdem ich mir an der Vanderbilt University das Thema „Advanced Big Data Processing Methods“ angehört hatte, war ich mir sicher, dass es ein Hut war. Möglicherweise dauert es ein oder zwei Stunden, um den Hive-Server so zu konfigurieren, dass er alle Daten durchläuft und über das Ergebnis berichtet. Da unsere Daten in AWS S3 gespeichert sind, habe ich den
Athena- Dienst verwendet, mit dem Sie Hive SQL-Abfragen auf S3-Daten anwenden können. Sie müssen den Hive-Cluster nicht konfigurieren / erhöhen und müssen nur für die gesuchten Daten bezahlen.
Nachdem ich Athena meine Daten und ihr Format gezeigt hatte, führte ich einige Tests mit ähnlichen Abfragen durch:
select * from intensityData limit 10;
Und schnell gut strukturierte Ergebnisse erhalten. Fertig.
Bis wir versuchten, die Daten in der Arbeit zu verwenden ...
Ich wurde gebeten, alle SNP-Informationen abzurufen, um das Modell darauf zu testen. Ich habe eine Abfrage ausgeführt:
select * from intensityData where snp = 'rs123456';
... und wartete. Nach acht Minuten und mehr als 4 TB der angeforderten Daten erhielt ich das Ergebnis. Athena berechnet eine Gebühr für die gefundene Datenmenge von 5 USD pro Terabyte. Diese einzelne Anfrage kostete also 20 US-Dollar und acht Minuten Wartezeit. Um das Modell nach allen Daten laufen zu lassen, musste man 38 Jahre warten und 50 Millionen Dollar zahlen. Offensichtlich passte dies nicht zu uns.
Es war notwendig, Parkett zu verwenden ...
Was ich gelernt habe : Seien Sie vorsichtig mit der Größe Ihrer Parkettdateien und ihrer Organisation.
Zuerst habe ich versucht, die Situation zu korrigieren, indem ich alle TSVs in
Parkettdateien konvertiert habe . Sie eignen sich für die Arbeit mit großen Datenmengen, da die darin enthaltenen Informationen in Spaltenform gespeichert sind: Im Gegensatz zu Textdateien, in denen Zeilen Elemente jeder Spalte enthalten, befindet sich jede Spalte in einem eigenen Speicher- / Festplattensegment. Und wenn Sie etwas finden müssen, lesen Sie einfach die erforderliche Spalte. Darüber hinaus wird in jeder Datei in einer Spalte ein Wertebereich gespeichert. Wenn der gewünschte Wert nicht im Spaltenbereich liegt, verschwendet Spark keine Zeit mit dem Scannen der gesamten Datei.
Ich habe eine einfache
AWS Glue- Aufgabe ausgeführt, um unsere TSVs in Parkett umzuwandeln, und neue Dateien in Athena abgelegt. Es dauerte ungefähr 5 Stunden. Aber als ich die Anfrage startete, dauerte es ungefähr die gleiche Zeit und etwas weniger Geld, um sie abzuschließen. Tatsache ist, dass Spark beim Versuch, die Aufgabe zu optimieren, einfach einen TSV-Block entpackt und in einen eigenen Parkettblock gelegt hat. Und da jeder Block groß genug war und die vollständigen Aufzeichnungen vieler Personen enthielt, wurden alle SNPs in jeder Datei gespeichert, sodass Spark alle Dateien öffnen musste, um die erforderlichen Informationen zu extrahieren.
Seltsamerweise ist der Standardkomprimierungstyp (und der empfohlene Komprimierungstyp) in Parkett - bissig - nicht aufteilbar. Daher blieb jeder Executor bei der Aufgabe, den gesamten 3,5-GB-Datensatz zu entpacken und herunterzuladen.

Wir verstehen das Problem
Was ich gelernt habe : Das Sortieren ist schwierig, besonders wenn die Daten verteilt sind.
Es schien mir, dass ich jetzt die Essenz des Problems verstand. Ich musste die Daten nur nach SNP-Spalten sortieren, nicht nach Personen. Dann werden mehrere SNPs in einem separaten Datenblock gespeichert, und dann manifestiert sich die intelligente Parkettfunktion „Nur öffnen, wenn der Wert im Bereich liegt“ in ihrer ganzen Pracht. Leider hat sich das Aussortieren von Milliarden von Zeilen, die über einen Cluster verteilt sind, als entmutigende Aufgabe erwiesen.
AWS möchte das Geld sicherlich nicht zurückgeben, weil "ich ein zerstreuter Student bin". Nachdem ich mit dem Sortieren auf Amazon Glue begonnen hatte, funktionierte es 2 Tage lang und stürzte ab.
Was ist mit Partitionierung?
Was ich gelernt habe : Partitionen in Spark sollten ausgeglichen sein.
Dann kam mir die Idee, die Daten auf den Chromosomen zu partitionieren. Es gibt 23 von ihnen (und einige weitere angesichts mitochondrialer DNA und nicht kartierter Bereiche).
Auf diese Weise können Sie die Daten in kleinere Teile aufteilen. Wenn Sie der Spark-Exportfunktion im Glue-Skript nur eine Zeile
partition_by = "chr" hinzufügen, sollten die Daten in Buckets sortiert werden.
 Das Genom besteht aus zahlreichen Fragmenten, die als Chromosomen bezeichnet werden.
Das Genom besteht aus zahlreichen Fragmenten, die als Chromosomen bezeichnet werden.Dies hat leider nicht funktioniert. Chromosomen haben unterschiedliche Größen und daher unterschiedliche Informationsmengen. Dies bedeutet, dass die Aufgaben, die Spark an die Mitarbeiter gesendet hat, nicht ausgeglichen und langsam ausgeführt wurden, da einige Knoten früher beendet wurden und inaktiv waren. Die Aufgaben wurden jedoch abgeschlossen. Bei der Anforderung eines SNP verursachte das Ungleichgewicht jedoch erneut Probleme. Die Kosten für die Verarbeitung von SNPs auf größeren Chromosomen (dh woher wir die Daten beziehen möchten) haben sich nur um das Zehnfache verringert. Viel, aber nicht genug.
Und wenn Sie sich in noch kleinere Partitionen aufteilen?
Was ich gelernt habe : Versuchen Sie niemals, 2,5 Millionen Partitionen zu erstellen.
Ich entschied mich für einen Spaziergang und teilte jeden SNP auf. Dies garantierte die gleiche Größe von Partitionen.
SCHLECHT WAR EINE IDEE . Ich nutzte Glue und fügte die unschuldige
partition_by = 'snp' . Die Aufgabe wurde gestartet und ausgeführt. Einen Tag später überprüfte ich, ob in S3 bisher nichts geschrieben war, und beendete die Aufgabe. Es sieht so aus, als hätte Glue Zwischendateien an einen versteckten Ort in S3 geschrieben, und viele Dateien, vielleicht ein paar Millionen. Infolgedessen kostete mein Fehler mehr als tausend Dollar und gefiel meinem Mentor nicht.
Partitionieren + Sortieren
Was ich gelernt habe : Das Sortieren ist immer noch schwierig, ebenso wie das Einrichten von Spark.
Der letzte Versuch der Partitionierung war, dass ich die Chromosomen partitionierte und dann jede Partition sortierte. Theoretisch würde dies jede Anforderung beschleunigen, da die gewünschten SNP-Daten innerhalb mehrerer Parkettblöcke innerhalb eines bestimmten Bereichs liegen sollten. Leider hat sich das Sortieren selbst partitionierter Daten als schwierige Aufgabe erwiesen. Infolgedessen wechselte ich für einen benutzerdefinierten Cluster zu EMR und verwendete acht leistungsstarke Instanzen (C5.4xl) und Sparklyr, um einen flexibleren Workflow zu erstellen ...
# Sparklyr snippet to partition by chr and sort w/in partition # Join the raw data with the snp bins raw_data group_by(chr) %>% arrange(Position) %>% Spark_write_Parquet( path = DUMP_LOC, mode = 'overwrite', partition_by = c('chr') )
... die Aufgabe wurde jedoch noch nicht erledigt. Ich habe in jeder Hinsicht optimiert: Ich habe die Speicherzuordnung für jeden Abfrage-Executor erhöht, Knoten mit einer großen Speichermenge verwendet, Broadcast-Variablen verwendet, aber jedes Mal stellte sich heraus, dass es sich um halbe Sachen handelte, und nach und nach versagten die Darsteller, bis alles aufhörte.
Ich werde erfinderischer
Was ich gelernt habe : Manchmal erfordern spezielle Daten spezielle Lösungen.
Jeder SNP hat einen Positionswert. Dies ist die Anzahl, die der Anzahl der Basen entspricht, die entlang des Chromosoms liegen. Dies ist eine gute und natürliche Art, unsere Daten zu organisieren. Zuerst wollte ich jedes Chromosom nach Regionen aufteilen. Zum Beispiel die Positionen 1 - 2000, 2001 - 4000 usw. Das Problem ist jedoch, dass SNPs nicht gleichmäßig über die Chromosomen verteilt sind, weshalb die Größe der Gruppen stark variiert.
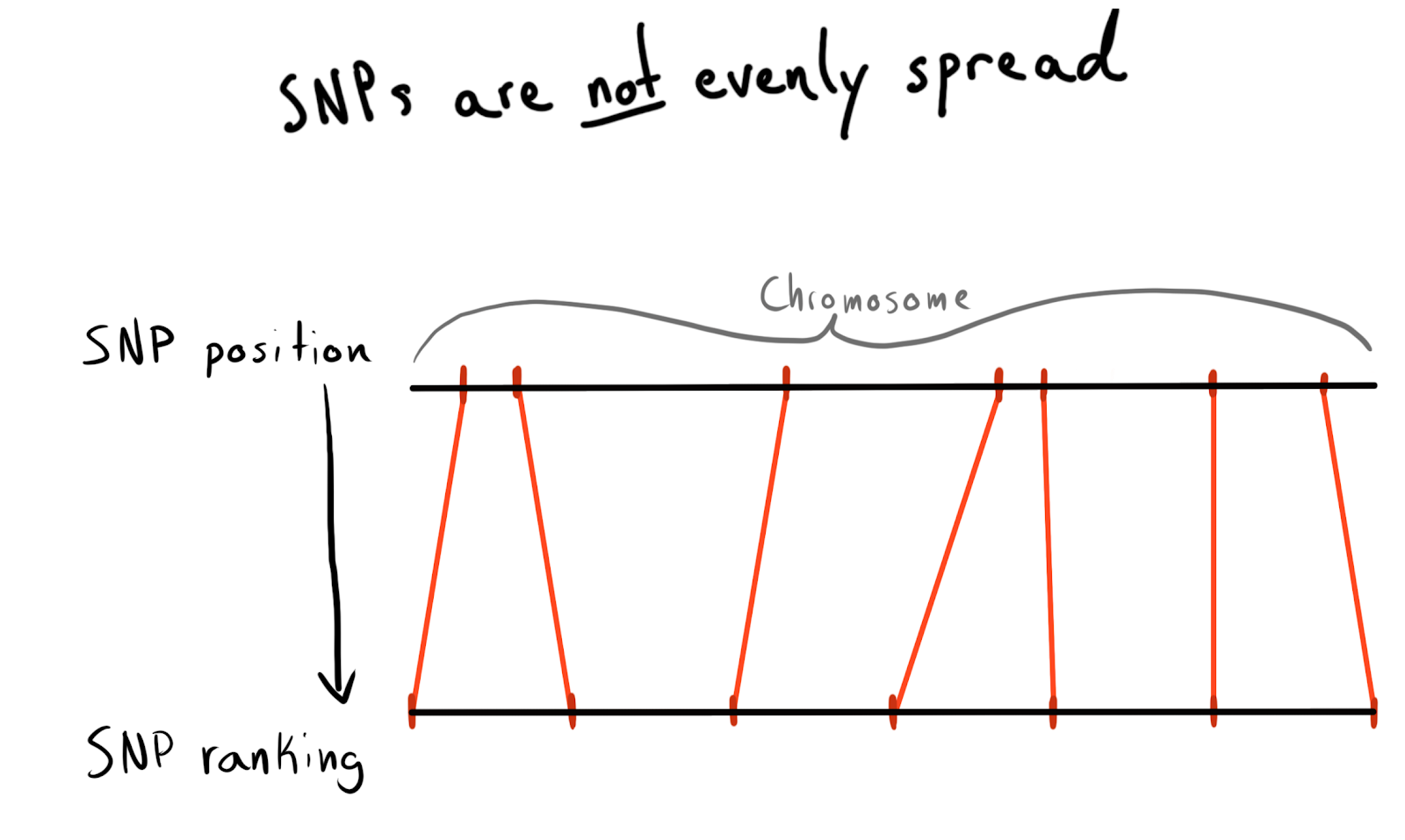
Infolgedessen wurde ich in Kategorien (Rang) Positionen eingeteilt. Gemäß den bereits heruntergeladenen Daten habe ich eine Liste mit eindeutigen SNPs, deren Positionen und Chromosomen angefordert. Dann sortierte er die Daten in jedem Chromosom und sammelte SNP in Gruppen (bin) einer bestimmten Größe. Sagen Sie jeweils 1000 SNP. Dies gab mir eine SNP-Beziehung zu einer Gruppe im Chromosom.
Am Ende habe ich Gruppen (bin) auf 75 SNP erstellt, ich werde den Grund unten erklären.
snp_to_bin <- unique_snps %>% group_by(chr) %>% arrange(position) %>% mutate( rank = 1:n() bin = floor(rank/snps_per_bin) ) %>% ungroup()
Versuchen Sie es zuerst mit Spark
Was ich gelernt habe : Die Spark-Integration ist schnell, aber die Partitionierung ist immer noch teuer.
Ich wollte diesen kleinen Datenrahmen (2,5 Millionen Zeilen) in Spark lesen, ihn mit Rohdaten kombinieren und dann durch die neu hinzugefügte
bin Spalte partitionieren.
Ich habe
sdf_broadcast() , damit Spark herausfindet, dass ein
sdf_broadcast() an alle Knoten
sdf_broadcast() werden soll. Dies ist nützlich, wenn die Daten klein sind und für alle Aufgaben benötigt werden. Andernfalls versucht Spark, intelligent zu sein und Daten nach Bedarf zu verteilen, was zu Bremsen führen kann.
Und wieder funktionierte meine Idee nicht: Die Aufgaben funktionierten eine Weile, schlossen die Fusion ab und begannen dann, wie die durch Partitionierung gestarteten Executoren, zu scheitern.
AWK hinzufügen
Was ich gelernt habe : Schlafen Sie nicht, wenn die Grundlagen es Ihnen beibringen. Sicherlich hat jemand Ihr Problem bereits in den 1980er Jahren gelöst.
Bis zu diesem Zeitpunkt war die Ursache all meiner Fehler mit Spark die Verwirrung der Daten im Cluster. Vielleicht kann die Situation durch Vorverarbeitung verbessert werden. Ich beschloss, die Rohtextdaten in Chromosomenspalten aufzuteilen, und hoffte, Spark mit „vorpartitionierten“ Daten versorgen zu können.
Ich habe in StackOverflow nach Möglichkeiten zum Aufteilen von Spaltenwerten gesucht und eine
so gute Antwort gefunden. Mit AWK können Sie eine Textdatei in Spaltenwerte aufteilen, indem Sie in das Skript schreiben, anstatt die Ergebnisse an
stdout senden.
Zum Testen habe ich ein Bash-Skript geschrieben. Ich habe einen der gepackten TSVs heruntergeladen, ihn dann mit
gzip entpackt und an
awk gesendet.
gzip -dc path/to/chunk/file.gz | awk -F '\t' \ '{print $1",..."$30">"chunked/"$chr"_chr"$15".csv"}'
Es hat funktioniert!
Kernfüllung
Was ich gelernt habe :
gnu parallel ist eine magische Sache, jeder sollte sie benutzen.
Die Trennung war ziemlich langsam, und als ich
htop startete, um die Verwendung einer leistungsstarken (und teuren) EC2-Instanz zu testen, stellte sich heraus, dass ich nur einen Kern und ungefähr 200 MB Speicher verwendete. Um das Problem zu lösen und nicht viel Geld zu verlieren, musste herausgefunden werden, wie die Arbeit parallelisiert werden kann. Glücklicherweise fand ich in Jeron Janssens 'beeindruckendem
Data Science at the Command Line- Buch ein Kapitel über Parallelisierung. Daraus lernte ich
gnu parallel , eine sehr flexible Methode zur Implementierung von Multithreading unter Unix.
Als ich die Partition mit einem neuen Prozess startete, war alles in Ordnung, aber es gab einen Engpass - das Herunterladen von S3-Objekten auf die Festplatte war nicht zu schnell und nicht vollständig parallelisiert. Um dies zu beheben, habe ich Folgendes getan:
- Ich fand heraus, dass es möglich ist, den S3-Download-Schritt direkt in der Pipeline zu implementieren, wodurch der Zwischenspeicher auf der Festplatte vollständig entfällt. Dies bedeutet, dass ich das Schreiben von Rohdaten auf die Festplatte vermeiden und noch kleineren und daher günstigeren Speicher auf AWS verwenden kann.
- Der Befehlssatz
aws configure set default.s3.max_concurrent_requests 50 hat die Anzahl der von der AWS CLI verwendeten Threads erheblich erhöht (standardmäßig sind es 10).
- Ich wechselte zu der für die Netzwerkgeschwindigkeit optimierten EC2-Instanz mit dem Buchstaben n im Namen. Ich fand heraus, dass der Verlust an Rechenleistung bei Verwendung von n-Instanzen durch eine Erhöhung der Download-Geschwindigkeit mehr als ausgeglichen wird. Für die meisten Aufgaben habe ich c5n.4xl verwendet.
- Ich habe
gzip in pigz geändert. Dies ist ein gzip-Tool, das coole Dinge tun kann, um die anfangs beispiellose Aufgabe des Entpackens von Dateien zu parallelisieren (dies hat am wenigsten geholfen).
Diese Schritte werden miteinander kombiniert, so dass alles sehr schnell funktioniert. Dank der erhöhten Download-Geschwindigkeit und der Ablehnung des Schreibens auf die Festplatte konnte ich jetzt ein 5-Terabyte-Paket in nur wenigen Stunden verarbeiten.
Dieser Tweet sollte "TSV" erwähnen. Leider.
Neu analysierte Daten verwenden
Was ich gelernt habe : Spark liebt unkomprimierte Daten und kombiniert keine Partitionen.
Jetzt waren die Daten in S3 in einem entpackten (gelesenen, freigegebenen) und halb geordneten Format, und ich konnte wieder zu Spark zurückkehren. Eine Überraschung erwartete mich: Ich habe wieder nicht das Gewünschte erreicht! Es war sehr schwierig, Spark genau zu sagen, wie die Daten partitioniert wurden. Und selbst als ich dies tat, stellte sich heraus, dass es zu viele Partitionen gab (95.000), und als ich ihre Anzahl mit
coalesce auf kohärente Grenzen reduzierte, ruinierte dies meine Partitionierung. Ich bin sicher, dass dies behoben werden kann, aber in ein paar Tagen der Suche konnte ich keine Lösung finden. Am Ende habe ich alle Aufgaben in Spark erledigt, obwohl es einige Zeit gedauert hat, und meine geteilten Parkettdateien waren nicht sehr klein (~ 200 Kb). Die Daten waren jedoch dort, wo sie benötigt wurden.
 Zu klein und anders, wunderbar!
Zu klein und anders, wunderbar!Testen lokaler Spark-Anforderungen
Was ich gelernt habe : Spark hat zu viel Aufwand bei der Lösung einfacher Probleme.
Durch das Herunterladen der Daten in einem intelligenten Format konnte ich die Geschwindigkeit testen. Ich habe ein Skript auf R eingerichtet, um den lokalen Spark-Server zu starten, und dann den Spark-Datenrahmen aus dem angegebenen Repository der Parkettgruppen (bin) geladen. Ich habe versucht, alle Daten zu laden, konnte Sparklyr jedoch nicht dazu bringen, die Partitionierung zu erkennen.
sc <- Spark_connect(master = "local") desired_snp <- 'rs34771739' # Start a timer start_time <- Sys.time() # Load the desired bin into Spark intensity_data <- sc %>% Spark_read_Parquet( name = 'intensity_data', path = get_snp_location(desired_snp), memory = FALSE ) # Subset bin to snp and then collect to local test_subset <- intensity_data %>% filter(SNP_Name == desired_snp) %>% collect() print(Sys.time() - start_time)
Die Ausführung dauerte 29.415 Sekunden. Viel besser, aber nicht zu gut für Massentests. Außerdem konnte ich die Arbeit mit dem Caching nicht beschleunigen, da Spark beim Versuch, den Datenrahmen im Speicher zwischenzuspeichern, immer abstürzte, selbst wenn ich mehr als 50 GB Speicher für ein Dataset mit einem Gewicht von weniger als 15 zugewiesen habe.
Kehre zu AWK zurück
Was ich gelernt habe : AWK assoziative Arrays sind sehr effizient.
Ich verstand, dass ich eine höhere Geschwindigkeit erreichen konnte. Ich erinnerte mich, dass ich in
Bruce Barnetts ausgezeichnetem AWK-Handbuch über eine coole Funktion namens "
Assoziative Arrays " gelesen habe. Tatsächlich handelt es sich hierbei um Schlüssel-Wert-Paare, die aus irgendeinem Grund in AWK anders bezeichnet wurden, und deshalb habe ich sie irgendwie nicht besonders erwähnt.
Roman Cheplyaka erinnerte daran, dass der Begriff „assoziative Arrays“ viel älter ist als der Begriff „Schlüssel-Wert-Paar“. Selbst wenn Sie
in Google Ngram nach Schlüsselwerten suchen , wird dieser Begriff dort nicht
angezeigt , aber Sie finden assoziative Arrays! Darüber hinaus wird das Schlüssel-Wert-Paar am häufigsten mit Datenbanken verknüpft, sodass ein Vergleich mit Hashmap viel logischer ist. Ich erkannte, dass ich diese assoziativen Arrays verwenden konnte, um meine SNPs mit der Bin-Tabelle und den Rohdaten zu verbinden, ohne Spark zu verwenden.
Dafür habe ich im AWK-Skript den
BEGIN Block verwendet. Dies ist ein Code, der ausgeführt wird, bevor die erste Datenzeile an den Hauptteil des Skripts übertragen wird.
join_data.awk BEGIN { FS=","; batch_num=substr(chunk,7,1); chunk_id=substr(chunk,15,2); while(getline < "snp_to_bin.csv") {bin[$1] = $2} } { print $0 > "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv" }
Mit dem Befehl
while(getline...) alle Zeilen aus der CSV-Gruppe (bin) geladen, die erste Spalte (SNP-Name) als Schlüssel für das assoziative Array
bin und der zweite Wert (group) als Wert festgelegt. Dann wird in dem Block
{ } , der auf alle Zeilen der Hauptdatei angewendet wird, jede Zeile an die Ausgabedatei gesendet, die abhängig von ihrer Gruppe (bin) einen eindeutigen Namen erhält:
..._bin_"bin[$1]"_...Die
chunk_id batch_num und
chunk_id entsprachen den von der Pipeline bereitgestellten Daten, wodurch der Race-Status vermieden wurde, und jeder
parallel gestartete Ausführungsthread schrieb in seine eigene eindeutige Datei.
Da ich alle Rohdaten in Ordnern auf den Chromosomen verteilt habe, die nach meinem vorherigen Experiment mit AWK übrig geblieben waren, konnte ich jetzt ein weiteres Bash-Skript schreiben, um es gleichzeitig auf dem Chromosom zu verarbeiten und S3 tiefer partitionierte Daten zu geben.
DESIRED_CHR='13'
Das Skript besteht aus zwei
parallel Abschnitten.
Im ersten Abschnitt werden Daten aus allen Dateien gelesen, die Informationen zum gewünschten Chromosom enthalten. Anschließend werden diese Daten auf Streams verteilt, die Dateien in die entsprechenden Gruppen (bin) verteilen. Um zu verhindern, dass Rennbedingungen auftreten, wenn mehrere Streams in eine einzelne Datei geschrieben werden, überträgt AWK die Dateinamen zum Schreiben von Daten an verschiedene Stellen, z. B.
chr_10_bin_52_batch_2_aa.csv . Infolgedessen werden viele kleine Dateien auf der Festplatte erstellt (dafür habe ich Terabyte-EBS-Volumes verwendet).
Die Pipeline aus dem zweiten
parallel Abschnitt durchläuft die Gruppen (bin) und kombiniert ihre einzelnen Dateien zu gemeinsamen CSVs mit
cat und sendet sie dann zum Export.
Sendung an R?
Was ich gelernt habe : Sie können über ein R-Skript auf
stdin und
stdout zugreifen und es daher in der Pipeline verwenden.
Im Bash-Skript stellen Sie möglicherweise folgende Zeile fest:
...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... Es übersetzt alle verketteten Gruppendateien (bin) in das unten stehende R-Skript.
{} ist eine spezielle
parallel Technik, bei der alle von ihr gesendeten Daten in den angegebenen Stream direkt in den Befehl selbst eingefügt werden. Die Option
{#} bietet eine eindeutige Thread-ID, und
{%} für die Job-Slot-Nummer (wiederholt, jedoch niemals gleichzeitig). Eine Liste aller Optionen finden Sie in der
Dokumentation. #!/usr/bin/env Rscript library(readr) library(aws.s3) # Read first command line argument data_destination <- commandArgs(trailingOnly = TRUE)[1] data_cols <- list(SNP_Name = 'c', ...) s3saveRDS( read_csv( file("stdin"), col_names = names(data_cols), col_types = data_cols ), object = data_destination )
Wenn die
readr::read_csv file("stdin") an
readr::read_csv , werden die in das R-Skript übersetzten Daten in den Frame geladen, der dann mit
aws.s3 als
.rds Datei direkt in S3
aws.s3 .
RDS ist ein bisschen wie eine jüngere Version von Parkett, ohne den Schnickschnack der Säulenlagerung.
Nach Abschluss des Bash-Skripts erhielt ich eine
.rds Dateien in S3, mit denen ich effiziente Komprimierung und integrierte Typen verwenden konnte.
Trotz der Verwendung der Bremse R funktionierte alles sehr schnell. Es ist nicht überraschend, dass die Fragmente auf R, die für das Lesen und Schreiben von Daten verantwortlich sind, gut optimiert sind. Nach dem Testen auf einem mittelgroßen Chromosom war die Aufgabe auf der C5n.4xl-Instanz in etwa zwei Stunden abgeschlossen.
S3 Einschränkungen
Was ich gelernt habe : Dank der intelligenten Implementierung von Pfaden kann S3 viele Dateien verarbeiten.
Ich war besorgt, ob S3 viele darauf übertragene Dateien verarbeiten könnte. Ich könnte die Dateinamen aussagekräftig machen, aber wie wird S3 danach suchen?
 S3 ,
S3 , / . FAQ- S3., S3 - . (bucket) , — .
Amazon, , «-----» . : get-, . , 20 . bin-. , , (, , ). .
?
: — .
: « ?» ( gzip CSV- 7 ) . , R Parquet ( Arrow) Spark. R, , , .
: , .
, .
EC2 , ( , Spark ). , , AWS- 10 .
R .
S3 , .
library(aws.s3) library(tidyverse) chr_sizes <- get_bucket_df( bucket = '...', prefix = '...', max = Inf ) %>% mutate(Size = as.numeric(Size)) %>% filter(Size != 0) %>% mutate(
, , ,
num_jobs , .
num_jobs <- 7
purrr .
1:1000 %>% map_df(shuffle_job) %>% filter(sd == min(sd)) %>% pull(data) %>% pluck(1)
, . Bash-
for . 10 . , . , .
for DESIRED_CHR in "16" "9" "7" "21" "MT" do
:
sudo shutdown -h now
… ! AWS CLI
user_data Bash- . , .
aws ec2 run-instances ...\ --tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]" \ --user-data file://<<job_script_loc>>
!
: API .
- . , . API .
.rds Parquet-, , . R-.
, ,
get_snp .
pkgdown , .
: , !
SNP , (binning) . SNP, (bin). ( ) .
, . , . ,
dplyr::filter , , .
,
prev_snp_results snps_in_bin . SNP (bin), , . SNP (bin) :
Ergebnisse
( ) , . , . .
, , , …
. . ( ), , (bin) , SNP 0,1 , , S3 .
Fazit
— . , . , . , , , . , , , , . , , , , - .
. , , «» , . .
:
- 25 ;
- Parquet- ;
- Spark ;
- 2,5 ;
- , Spark;
- ;
- Spark , ;
- , , - 1980-;
gnu parallel — , ;
- Spark ;
- Spark ;
- AWK ;
stdin stdout R-, ;
- S3 ;
- — ;
- , ;
- API ;
- , !