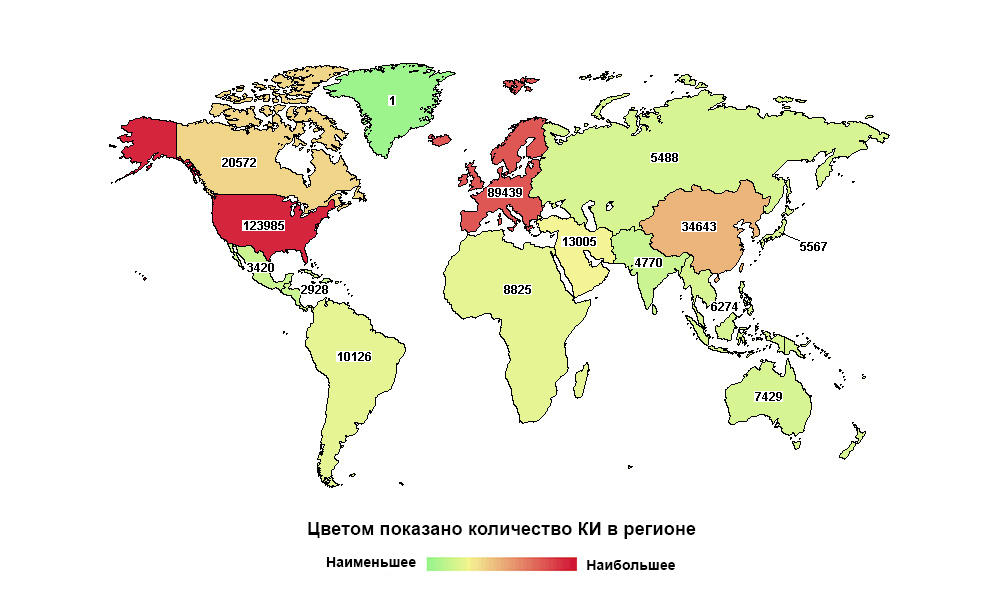
 Russland ist weit davon entfernt, das erste der Welt zu sein, aber das erste in der Anzahl der Studien in seiner Makroregion
Russland ist weit davon entfernt, das erste der Welt zu sein, aber das erste in der Anzahl der Studien in seiner MakroregionJedes Medikament durchläuft heute, bevor es zum Patienten gelangt, eine lange Reihe klinischer Studien. Es muss nachgewiesen werden, dass es in der Lage ist, ein bestimmtes Gesundheitsproblem zu lösen, und dies effektiver und vorzugsweise sicherer als seine Vorgänger.
Die Auswahl ist eng - 98% aller untersuchten Medikamente erreichen die Patienten nicht. Bei 2% der „Glücklichen“ dauert die wissenschaftliche Erforschung einer neuen Substanz vor dem Markteintritt mehr als 12 Jahre und mehr als 1,5 Milliarden Dollar.
Wir von der
Medicine 24/7 Clinic sind direkt an klinischen Studien beteiligt. Zum zweiten Mal in Folge führen wir klinische Studien mit ausländischen Antitumormitteln durch. Mit unserer Hilfe erhalten neue Medikamente einen schnelleren Zugang zu Russland und mehr als 100 Menschen pro Jahr - eine weitere kostenlose Behandlungschance.
Für Privatkliniken ist die Praxis ungewöhnlich: ein Minimum an kommerziellen Vorteilen, zu viele Schwierigkeiten bei der Organisation des Prozesses und strenge Anforderungen an eine medizinische Einrichtung. In der Regel schaffen es nur große Bundeszentren, diese einzuhalten.
Für viele Patienten in Russland ist eine klinische Prüfung des Arzneimittels jedoch die einzige Möglichkeit, eine kostenlose Behandlung für eine tödliche Krankheit zu erhalten. Aber unter den russischen Krebspatienten wissen 30% einfach nicht, was eine klinische Studie ist, und nur wenige haben an ihnen teilgenommen.
Deshalb möchten wir, dass so viele Menschen wie möglich lernen und prüfen: Vielleicht haben sie die Chance, ein Medikament zu bekommen, das ihr Leben retten kann.
In diesem Artikel erklären wir, warum wir brauchen und wie klinische Studien organisiert sind, wer und wie wir dorthin gelangen können.
Traurige Geschichten. Warum sind klinische Studien erforderlich und warum ist ohne sie schlecht
Klinische Forschung / Studie (im Folgenden: CI) - eine wissenschaftliche Studie, an der Menschen als Probanden beteiligt sind, um die Wirksamkeit und Sicherheit eines neuen Arzneimittels zu bewerten oder die Indikationen für die Verwendung des bereits bekannten Arzneimittels zu erweitern. Neben Medikamenten können CIs auch die Wirksamkeit und Sicherheit neuer Behandlungs- und Diagnosemethoden untersuchen.
Die Medizin entwickelt sich und entwickelt sich zu einer exakten Wissenschaft, die ohne Statistik nicht auskommt.
Zuvor kannte der Hausarzt die Geschichten aller seiner Patienten auswendig, der Arzt konnte sein ganzes Leben in einer Stadt verbringen, einen persönlichen Ansatz zur Behandlung aller finden und sich daran erinnern. Darüber hinaus war die Auswahl an Tränken gering: Heilkräuter, Blutegel, Quecksilber und Arsen. Die Verantwortung während des Postulats "allen Willens Gottes" gegenüber den Ärzten war geringer.
 Arsen vom Ende des 18. Jahrhunderts "wiederhergestellte" Potenz und "geheilte" Arthritis ...
Arsen vom Ende des 18. Jahrhunderts "wiederhergestellte" Potenz und "geheilte" Arthritis ... ... und Quecksilber zum Beispiel war ein Abführmittel und "von Syphilis".
... und Quecksilber zum Beispiel war ein Abführmittel und "von Syphilis".Als sich die Medizin verbreitete, mussten die Ärzte wirklich unverwechselbare Behandlungstaktiken entwickeln. Bestimmte Medikamente sollten den meisten Patienten unter bestimmten Bedingungen helfen.
Im Idealfall sollte ein Arzt nur solche Methoden zur Vorbeugung, Diagnose und Behandlung anwenden, bei denen die Wahrscheinlichkeit, dass „zufällige Ergebnisse“ erzielt werden, äußerst gering ist, da ihre Nützlichkeit und Wirksamkeit durch viele korrekt durchgeführte Experimente nachgewiesen wurde.
Dies ist
evidenzbasierte Medizin - der einzig angemessene Ansatz für eine so ernste Angelegenheit wie die heutige menschliche Gesundheit.
Und es ist die
klinische Forschung, die die
Grundlage der evidenzbasierten Medizin bildet.
Bis zur Mitte des 20. (!) Jahrhunderts gab es keine Regulierung der Forschung zu neuen Arzneimitteln. Wie so oft dauerte es einige Tragödien, um die Ordnung wiederherzustellen.
1937 starben 105 Kinder und ein Erwachsener und nahmen das "Elixier" aus dem Antiseptikum Sulfonamid und ... giftigem Diethylenglykol. Ja, die, die heute in Frostschutzmitteln verwendet wird. Dann verwendete das Pharmaunternehmen
ME Massengill es unwissentlich als Lösungsmittel, Hilfsstoff. Es wurden keine Sicherheitsstudien des resultierenden „Cocktails“ für den Menschen durchgeführt. Als sie plötzlich bemerkten und die Droge aus dem Verkauf nahmen, gab es bereits mehr als hundert Opfer. 1938 verabschiedete der US-Kongress ein Gesetz über die obligatorische Erforschung von Arzneimitteln, bevor sie in den Handel kommen. Die Kontrolle darüber wurde der FDA (
English Food and Drug Administration), der Food and Drug Administration, übertragenEin noch lauterer Skandal ereignete sich Ende der 1950er und Anfang der 1960er Jahre mit
Thalidomid . "Beruhigende und schlafende Pillen, die perfekt bei der Toxikose schwangerer Frauen helfen" waren schnell ausverkauft. Seine Studien wurden nur an Ratten durchgeführt. Es stellte sich heraus, dass dies nicht genug ist. Beim Menschen verursachte Thalidomid Defekte in der Entwicklung des Fötus. In Europa, Australien und Japan wurden etwa 10.000 Kinder mit Missbildungen (Missbildungen) der Gliedmaßen geboren. Das Medikament wurde 1961 in den meisten Ländern verboten.
 Mütter dieser Kinder tranken Schlaftabletten, die nicht an Menschen getestet wurden
Mütter dieser Kinder tranken Schlaftabletten, die nicht an Menschen getestet wurdenSeitdem wurden Arzneimittel vor der Registrierung sorgfältig untersucht. Dies wird durch die
International Harmonized Tripartite Rules of Good Clinical Practice (Harmonisierte dreigliedrige ICH-Richtlinie für gute klinische Praxis, abgekürzt als ICH GCP) geregelt
. Von 1996 bis 1997 sind sie in den USA, Japan und der EU tätig und werden seit 2003 in Russland eingeführt.Wie läuft die Studie und warum so lange?
Der gesamte Prozess der Herstellung des Arzneimittels kann in drei große Phasen unterteilt werden.
1. Suche nach Ideen und präklinischen Studien - in vitro und bei Tieren.
2. Wenn dies nicht dort endete, beginnen klinische Studien mit Menschen: zuerst vorsichtig, dann massiver.
3. Das Medikament wird dann bei den Aufsichtsbehörden registriert, um in medizinischen Verzeichnissen ein bekannter Name zu werden.
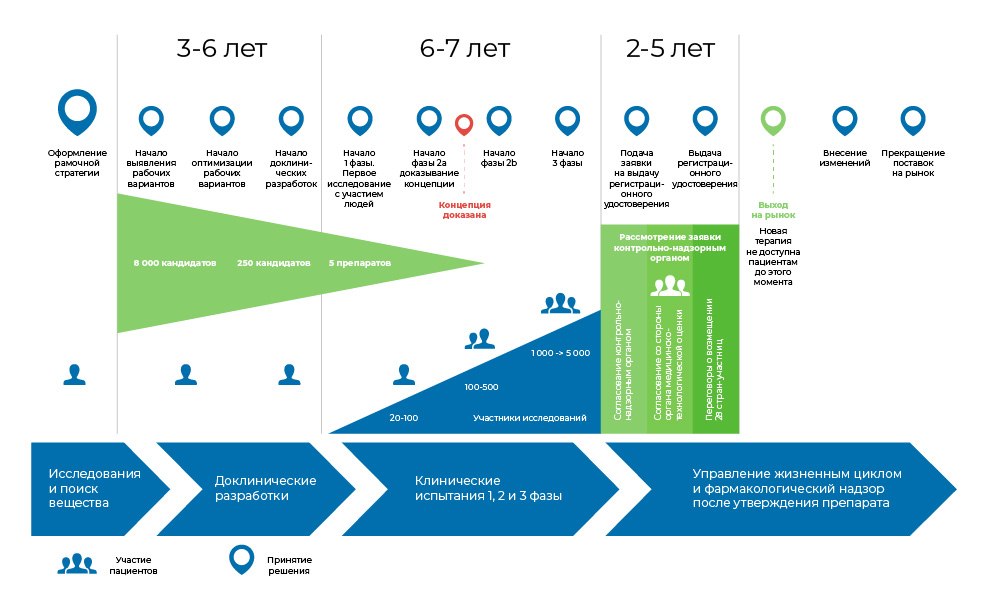 Der Prozess der Entwicklung eines Arzneimittels. Von der Entstehung des Moleküls bis zum Beginn des Verkaufs des Arzneimittels dauert es 8 bis 20 Jahre.Braucht das jemand?
Der Prozess der Entwicklung eines Arzneimittels. Von der Entstehung des Moleküls bis zum Beginn des Verkaufs des Arzneimittels dauert es 8 bis 20 Jahre.Braucht das jemand? Die Onkologie ist einer der ungeheuerlichsten Bereiche der Medizin im Hinblick auf den ungedeckten Bedarf an Medikamenten. Laut der Weltgesundheitsorganisation
starben 2018 9,6 Millionen Menschen an Krebs. Tumore treten häufig in späteren Stadien auf, wenn nur noch eine palliative Behandlung verbleibt.
Gleichzeitig ermöglichten Entdeckungen auf dem Gebiet der Molekularbiologie und Genetik, die Mechanismen zu verstehen, die zur Entwicklung und zum Fortschreiten von Krebs beitragen, und das Verständnis der Arbeit der Antitumorimmunität zu verbessern.
Und heute ist die Entwicklung von Antitumormitteln eines der wissenschaftsintensivsten und beliebtesten Gebiete der Medizin.
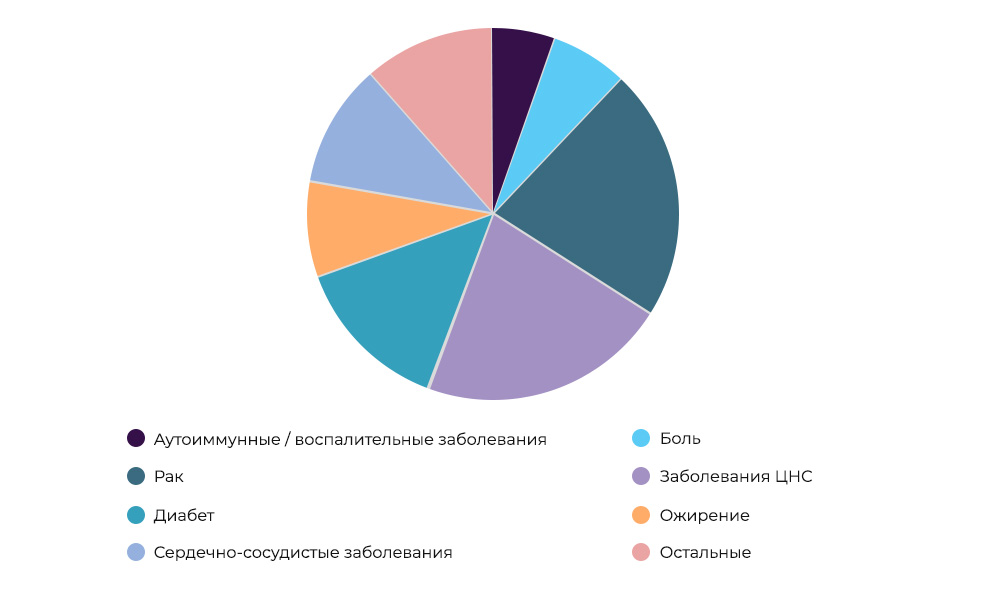 Forschung zu Krebsmedikamenten - 23% aller CI weltweitWer bezahlt die Forschung.
Forschung zu Krebsmedikamenten - 23% aller CI weltweitWer bezahlt die Forschung. Manchmal kann der Organisator und Sponsor eine Forschungsorganisation sein. Aber häufiger betreiben Wissenschaftler wissenschaftliche Forschung auf Kosten von Pharmaunternehmen. Diese erwarten, mit einem erfolgreichen Medikament auf den Markt zu kommen, Gewinne zu erzielen und die Kosten für CI und Entwicklung wieder hereinzuholen. Es ist wie beim Kauf eines neuen Films zum Ausleihen in einem Film: Der Verleiher weiß nicht, ob er "drehen" wird oder nicht. Die Herstellung neuer Medikamente ist ein sehr riskantes Geschäft.
Zuvor forschten viele Pharmaunternehmen allein mit Wissenschaftlern. Jetzt kann eine medizinische Einrichtung, die die Akkreditierung bestanden hat und bestimmte Anforderungen erfüllt, zur Plattform und Durchführung des Experiments werden.
Genau das ist bei unserer
„Medizin 24/7“ der Fall. Das Pharmaunternehmen ist bereit zu zahlen, aber die Finanzen werden am Ende der Studie auf der Grundlage der angefallenen Kosten zugewiesen. Die Klinik verdient keine Superprofits. Und Forschungsärzte verdienen in der Regel nichts zusätzlich zu ihrem üblichen Gehalt. Dies ist vielmehr die Position des Klinikleiters: Eine Person hält es für richtig, die Medizin des Landes voranzubringen, und nutzt die Gelegenheit, sich daran zu beteiligen.
Zuerst kommt eine Idee auf. Was soll man eigentlich untersuchen? In der Onkologie werden zuerst „Ziele“ gefunden - eine Schwachstelle der Krankheit. Wenn Sie die Zielmoleküle stören oder einfach „ausschalten“, „leidet“ der Tumor. Viele moderne Krebsmedikamente,
gezielt oder
immuntherapeutisch, bauen auf diesem Prinzip auf.
 Der Mechanismus gezielter Medikamente bei Darmkrebs. Krebszellen teilen sich nicht mehr oder wachsen zusätzliche Blutgefäße zum Tumor, oder das Medikament schützt benachbarte Zellen davor, bösartig zu werden
Der Mechanismus gezielter Medikamente bei Darmkrebs. Krebszellen teilen sich nicht mehr oder wachsen zusätzliche Blutgefäße zum Tumor, oder das Medikament schützt benachbarte Zellen davor, bösartig zu werdenUm solche Substanzen zu finden und dann die am besten geeigneten Kandidaten auszuwählen, sind viele Ressourcen und Zeit für
In-vitro- und Silicio-Studien erforderlich, dh in vitro oder mithilfe von Computersimulationen.
Der ausgewählte Stoff wird in der richtigen Menge gelagert - hergestellt nach speziellen Regeln (in Russland ist es GOST R 52249-2009), ohne Verunreinigungen und Verstöße gegen die Technologie. Und mit diesen Reagenzgläsern testen Wissenschaftler das Medikament an Tieren.
Die Maus ist der Motor des Fortschritts. Nach dem Testen von Ideen in vitro geht ein Wissenschaftler mit einem Vorrat seines potenziellen Arzneimittels ins Vivarium. Sie müssen überprüfen, wie sich der Prototyp im Körper eines Säugetiers verhält (in vivo).
Bereits 1025 schrieb Avicenna im "Canon of Medical Science", dass Medikamente überprüft werden müssen. Darüber hinaus ist es wünschenswert - bei einem potenziellen Patienten, einer Person. Schließlich garantiert das Ergebnis bei Löwen und Pferden nicht, dass das Arzneimittel die Menschen in gleicher Weise beeinflusst.
Und noch in der Medizin, ohne Tierversuche - geht nicht. Die Löwen und Pferde wurden jedoch allein gelassen. Präklinische Studien auf der ganzen Welt finden hauptsächlich an Mäusen, Meerschweinchen und Kaninchen statt.
 Die Labormäuse haben sogar ein Denkmal in den Nowosibirsk Academgorodok gesetzt
Die Labormäuse haben sogar ein Denkmal in den Nowosibirsk Academgorodok gesetztÜberprüfen Sie zu diesem Zeitpunkt, wie schädlich / sicher das Medikament ist:
- Verursacht es Allergien?
- Hat es toxische Wirkungen auf Gewebe und Organe?
- wie es die Fortpflanzungsfähigkeit der Tiere und die normale Entwicklung des Fötus usw. beeinflusst
Darüber hinaus beobachten sie, wie sich der Medikamentenkandidat im Körper des Tieres verhält (Pharmakokinetik):
- Absorptionsrate und Erhöhung der Blutkonzentration,
- Was sind die maximale und minimale Dosis
- wie schnell es aus dem Körper ausgeschieden wird usw.
Alle diese Daten werden benötigt, um zu entscheiden,
ob es möglich ist, die Testsubstanz für den Menschen zu verwenden. Und wenn ja, wie viel wird benötigt?
Unvermeidliches Übel. Bürokratie Das
Außenministerium überwacht den korrekten Fortschritt von CI
. Regulierung des Arzneimittelverkehrs des Gesundheitsministeriums und des Bundesdienstes für die Überwachung des Gesundheitswesens (Roszdravnadzor).Wenn der Wissenschaftler zu dem Zeitpunkt gekommen ist, an dem klinische Studien am Menschen durchgeführt werden müssen, ist es an der Zeit, einen Antrag für die Durchführung von CI vorzubereiten. Dazu benötigt er mehrere
Dokumente .
- Dossier des Studienmedikaments. Alles, was bereits über das Medikament herausgefunden wurde: Daten zur Pharmakokinetik, Wirksamkeit, Toxizität usw.
- Studienprotokoll Es enthält Einzelheiten zum Plan für künftige Forschungsarbeiten und Methoden zur Bewertung der Ergebnisse.
- Forscherbroschüre. Ein kurzer Spickzettel, um Freiwilligen und Patienten das Wesentliche der Studie klar zu erklären und ihre Einverständniserklärung einzuholen.
Ethikkommission. Die nächste Phase der Suche besteht darin, die Bewertung und den Abschluss der Ethikkommission zu erhalten.
Die Ethikkommission ist eine unabhängige Gruppe von Ärzten, Wissenschaftlern, medizinischem Personal und Nichtfachleuten (Mitgliedern der Öffentlichkeit). Sie studieren das Studienprotokoll und die Einverständniserklärung, um sicherzustellen, dass zwischen dem Patienten, den Forschern, dem Pharmaunternehmen und der nationalen Regulierungsbehörde eine Einigung erzielt wurde, dass niemandes Rechte verletzt wurden, niemand einem Zwang ausgesetzt wurde und niemand gegen den freien Willen verstoßen hat.
Nach schwerwiegenden Nebenwirkungen eines Arzneimittels im Jahr 2006 wurde die Ethikkommission noch strenger. Daher kann es vorkommen, dass die Studie zu diesem Zeitpunkt für ein Jahr oder länger „einfriert“.
Nach Prüfung aller Dokumente und Genehmigung durch die Ethikkommission befindet sich das potenzielle Medikament in der Phase
klinischer Studien - beim Menschen.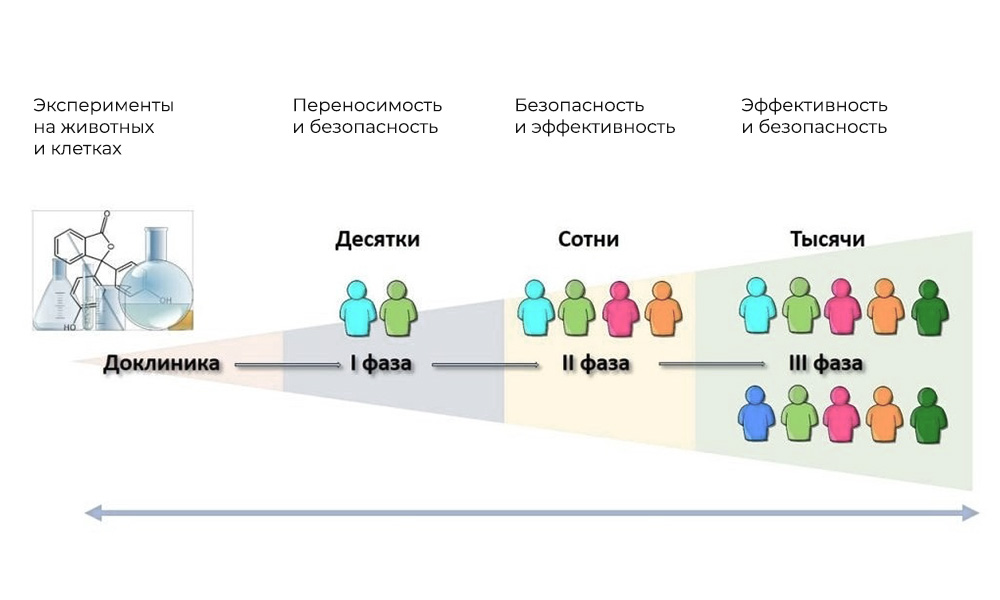 Die Hauptphasen klinischer Studien liegen beim Menschen.
Die Hauptphasen klinischer Studien liegen beim Menschen.Phase I. Testen des Wirkmechanismus
Teilnehmer: 20 - 100 Personen.
Dauer: von mehreren Monaten bis 1 Jahr.
Ziel: Untersuchung von Toleranz, Pharmakodynamik und Pharmakokinetik.
Es wird geprüft, ob der Stoff beim Menschen genauso wirkt wie bei Tieren, ob er sicher ist.
In der ersten Phase einer klinischen Studie sollten gesunde Freiwillige theoretisch teilnehmen, aber in der Onkologie kann das Testen wirksamer Substanzen in einem gesunden Körper nicht als ethisch bezeichnet werden. Daher sind Menschen mit der entsprechenden Krankheit beteiligt, gegen die das zukünftige Medikament wirksam sein kann.
Den Teilnehmern werden nach und nach alle größeren Dosen des Arzneimittels injiziert, beginnend mit dem minimal bis maximal zulässigen Wert. Nach jeder Verabreichung wird der Zustand des Patienten überwacht.
Die Pharmakokinetik wird bewertet : Absorptionsrate und Ausscheidung (Ausscheidung unveränderter Substanzen), Verteilung über Gewebe und Organe.
Die Pharmakodynamik wird ebenfalls bewertet: die Wirkung des Arzneimittels auf Tumorzellen, auf andere Organe und Organe, Nebenwirkungen. Die bevorzugte Anwendung und Dosierung werden geklärt.
Zusätzlich zu Studien mit steigenden Dosen wird in Phase I Folgendes überprüft:
- die Wirkung von Lebensmitteln auf das Medikament;
- Interaktion mit anderen Drogen;
- die Wirkung anderer Krankheiten, die die gewünschte Dosis des Arzneimittels beeinflussen können (z. B. bei einem Patienten mit Nierenversagen).
Laut
FDA bestehen 70% der Medikamente die erste Phase des CI erfolgreich.
Phase II Überprüfung der Aktion für ein bestimmtes Ziel: eine bestimmte Art von Krankheit
Teilnehmer: 100 bis 500 Patienten.
Dauer: von mehreren Monaten bis 2 Jahren.
Zweck: um die Wirksamkeit bestimmter Indikationen
zu testen
Es muss untersucht werden, wie wirksam das neue Medikament im Vergleich zu Placebo oder einer bestehenden Behandlung ist. Außerdem kann eine größere Anzahl von Teilnehmern seltenere Nebenwirkungen erkennen, die in Phase I nicht erkannt werden.
Um an dieser Phase der CI teilzunehmen, werden die Patienten nach einer viel größeren Anzahl von Kriterien ausgewählt als in der ersten Phase. Zum Beispiel nicht nur "Brustkrebs", sondern "Brustkrebs, Stadium T2N1M0, HER2-positiver Subtyp".
Typischerweise werden Studien in diesem Stadium als
doppelblinde, randomisierte, placebokontrollierte Studien durchgeführt
.Doppelblindheit: Weder der Arzt noch der Patient wissen, wer den Wirkstoff erhält und wer ein Placebo oder die derzeit bestehende optimale Behandlung erhält.
Randomisierung bedeutet, dass Patienten zufällig in Gruppen eingeteilt werden - unter Verwendung eines Zufallszahlengenerators. Weder der Arzt noch der CI-Teilnehmer können diesen Prozess beeinflussen.
Placebo-Kontrolle bedeutet, dass Teilnehmer einer Gruppe unter den gleichen Bedingungen ein Placebo erhalten wie Teilnehmer einer anderen Gruppe, denen der Wirkstoff verabreicht wird.
 Jeder - das gleiche Aussehen, Geschmack und Geruch der Medizin.
Jeder - das gleiche Aussehen, Geschmack und Geruch der Medizin.All diese „Verschwörungstheologie“ ist erforderlich, um eine absichtliche oder unbewusste Verfälschung experimenteller Daten durch Teilnehmer oder Forscher auszuschließen.
In der ersten Phase, in der es keine so strengen Anforderungen gibt, gibt es erstaunliche Ergebnisse. Dies ist eine „schmutzige“ Statistik, in Phase II wird sie von Überschuss befreit und die Ergebnisse werden plausibel.
Laut FDA durchlaufen nur 33% der Medikamente, die die Phase II erreichen, erfolgreich eine CI und gehen in die nächste Phase über.
Phase III Unterstützende Studien
Teilnehmerzahl: 300 - 3.000 oder mehr.
Dauer: von einem Jahr bis zu mehreren Jahren.
Zweck: Bestätigung der Wirksamkeit und Sicherheit der Testsubstanz in großen Proben.
Dies ist der größte, komplexeste und teuerste Teil des Arzneimittelentwicklungsprozesses. Der Zweck solcher Studien besteht darin, die Wirksamkeit und Sicherheit der Testsubstanz bei Verwendung durch eine große Anzahl von Patienten zu bestätigen.
Basierend auf den Ergebnissen dieser Phase erhalten Arzneimittelhersteller die Erlaubnis, sie auf den Markt zu bringen.
In Phase III können Tausende von Patienten aus verschiedenen Ländern teilnehmen. Alles sollte bis ins kleinste Detail geplant werden, damit an allen Stellen der Studie das Design und die wesentlichen Bedingungen genau gleich sind.
Das Design der Studie ist so eng, dass nicht nur ein sterbender Patient, sondern auch ein Patient mit einer Prognose für eine stabile Remission in die Studie eintreten kann. Das Medikament sollte so sicher sein, dass es einem praktisch gesunden Menschen verabreicht werden kann - und die Lebensqualität nimmt nicht ab.
Vor Beginn der Phase III finden zahlreiche Konsultationen und Diskussionen zwischen Forschern und Experten von Drittanbietern statt: Es ist sehr wichtig, über die Versuchsplanung nachzudenken, um das Wichtige nicht zu verpassen und alle erforderlichen Daten zu erhalten.
Während der Phase III werden die Wirksamkeit und Sicherheit des neuen Arzneimittels sowie die Dosis-Wirkungs-Beziehung endgültig bestätigt.
Die Korrelation von Vorteilen und Risiken wird analysiert. Auf der Grundlage der Ergebnisse entscheidet die Regulierungsbehörde, ob es möglich ist, das Arzneimittel auf den Markt zu bringen. Dazu müssen folgende Bedingungen erfüllt sein:
- das Medikament ist wirksamer als bisher bekannte Analoga,
- gibt weniger Nebenwirkungen / besser verträglich
- wirksam, wenn bisher bekannte Medikamente nicht wirken,
- wirtschaftlich rentabler,
- einfacher zu bedienen.
Der Überprüfungsprozess des Antrags durch den Vorgesetzten dauert 12 bis 18 Monate.
Nach Angaben der FDA endet die dritte Phase der klinischen Studien mit einem positiven Ergebnis in nur 25 bis 30% aller Fälle zu Beginn der dritten Phase.
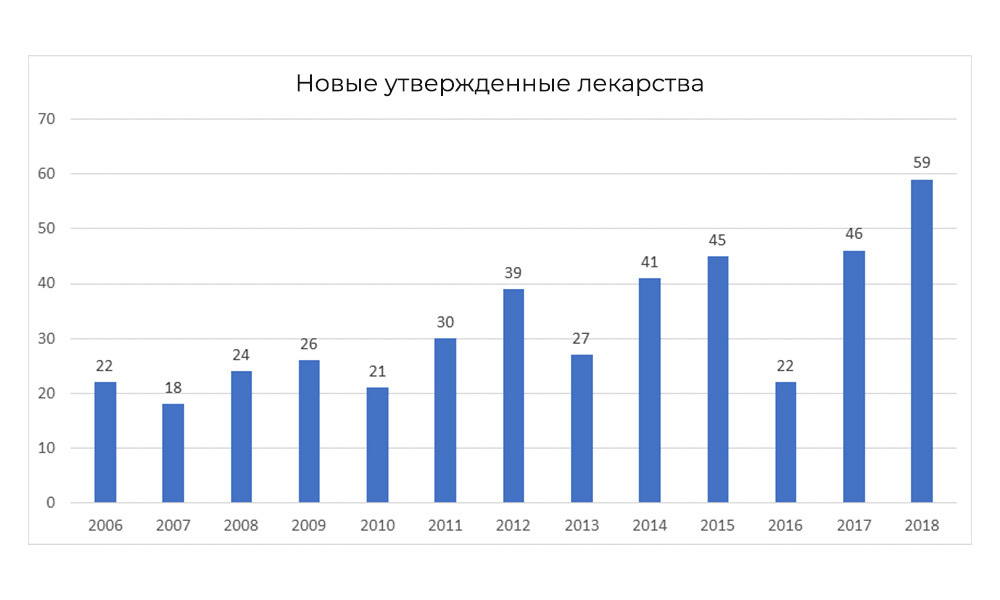 Im Jahr 2018 brach die FDA jedoch ihren eigenen Rekord für die Anzahl der zugelassenen Medikamente.
Im Jahr 2018 brach die FDA jedoch ihren eigenen Rekord für die Anzahl der zugelassenen Medikamente.Merkmale der nationalen Forschung: eine zusätzliche Phase des CI in Russland
Die Kontrolle neuer Medikamente in Russland hat ihre eigenen Fehler (oder Merkmale, wie sie aussehen sollen). Laut Gesetz müssen zugelassene ausländische Arzneimittel in Russland zusätzlichen klinischen Studien unterzogen werden. Dies soll die Qualität ausländischer Arzneimittel verbessern.
3 , , . , , , 3 , , .
, 12 – 12 , . , 6 12. , , , , .
2-3 .
« 24/7» III.
. , « ». , . , – , , .
, III – – , «» .
, . .
-,
GCP, Good Clinical Practice.-, . , - : - . . .
-, . , , , , , , – . – : , .
 –
–, , , , .
1 200 , . : , , , , . « » – - .
2 3 , .
2 :
. , , , , .. —
. .
– , , .
, . «», : , , / , – .
. , – . , .
18 .
, – , , 3-4 . : 10 , – .
, , .
– – . , . , , .
, , , , – – . .
, .
–
RosOncoWeb ,
CTAgency .
« 24/7» – .