Im vorherigen detaillierten
Artikel über das
vollständige Genom haben wir versprochen, drei Probleme zu veröffentlichen und demjenigen einen Test zu geben, der alle drei zuerst richtig löst. Gleichzeitig geben wir Beispiele, wie man bei diesen Aufgaben mit genetischen Daten arbeitet. Heute veröffentlichen wir die erste.

Im ersten
Artikel haben wir nützliche Informationen und Links geteilt, die für die Arbeit mit Bioinformatikdaten nützlich sind. Wir empfehlen, dass Sie es zuerst lesen, wenn Sie es verpasst haben.
HaftungsausschlussDie Arbeit mit genetischen Daten wird auf Unix-Systemen (Linux, macOS) ausgeführt, da einige Befehle und Software unter Windows nicht verfügbar sind. Aus diesem Grund besteht eine der einfachsten Lösungen für Windows-Benutzer darin, eine virtuelle Linux-Maschine zu mieten.
Alle nachfolgend beschriebenen Vorgänge werden auf dem Befehlszeilenterminal ausgeführt. Bevor Sie beginnen, lernen Sie, wie Sie in einem Terminal mit Ihrem Betriebssystem arbeiten und Befehle verwenden, da einige davon möglicherweise das Betriebssystem und Ihre Daten beschädigen können.
Benötigte Software
Wir haben das
Image einer virtuellen Maschine (VM) mit der gesamten erforderlichen Software auf Yandex.Cloud gesammelt. Registrieren Sie sich in Yandex.Cloud und klicken Sie in Ihrem Konto im Bereich "Compute Cloud" auf "VM erstellen". Wählen Sie als öffentliches Bild 1000 Genome aus dem Atlas Data Analysis-Katalog aus.
VM-Konfiguration: 100% 2vCPU, 8 GB RAM, 20 GB Festplatte. Beim Erstellen einer VM müssen Sie die Zahlungsdaten eingeben, es wird jedoch nichts vom Konto abgeschrieben. Ein Start und ein zusätzliches Grant für ein Codewort reichen aus, um mit einer VM und einem Image von Atlas bis zum 31. Dezember 2019 kostenlos zu arbeiten. Senden Sie das Codewort "ATLAS" an den
Yandex.Cloud-Support, um eine Finanzhilfe für die Erledigung von Aufgaben zu
erhalten .
Hinweis: Das Stipendium gilt für neue Yandex.Cloud-Benutzer, die sich seit dem 18. Dezember 2019 angemeldet haben, oder für Benutzer, die noch eine Testphase haben und ein Startstipendium haben. Das Codewort ATLAS ist nur einmal gültig.
Erstellen Sie zunächst einen SSH-Schlüssel auf dem lokalen Computer, von dem aus Sie eine Verbindung zur VM herstellen möchten:
ssh-keygen -o -t rsa -b 4096 -C "my-local-machine" -f ~/.ssh/yandex-cloud -a 100
Vergessen Sie nicht, den Inhalt der Datei
~/.ssh/yandex-cloud.pub beim Erstellen der VM in das entsprechende Fenster zu kopieren.
Wenn Sie die Software auf Ihrem Computer installieren möchten, finden Sie nachfolgend alle Installationsinformationen. Wenn Sie sich für Yandex.Cloud entscheiden, erstellen Sie eine VM und fahren Sie mit dem nächsten Abschnitt fort.
Plink
Plink ist ein Softwarepaket zur Manipulation genetischer Daten und der Wide Genome Association Search (GWAS). Es wurde vom Genetiker Sean Purcell (Shaun Purcell) entwickelt. Seit 2008 wurden mit Hilfe von Plink weltweit Hunderte von GWAS durchgeführt, deren beste Ergebnisse Atlas als Datenquelle für Algorithmen zur Berechnung von Krankheitsrisiken verwendet.
Plink bietet eine Reihe von Tools zum Speichern, Konvertieren und Suchen von Genotypisierungsdaten. Plink ermöglicht auch die statistische Verarbeitung, die Analyse des Verknüpfungsungleichgewichts (LD), die Analyse der Identität nach Abstammung (IBD) und der Identität (Zustand nach IBS), Populationsschichtungstests und Epistasietests - das Zusammenspiel mehrerer genetischer Variationen untereinander.
IBD und IBS werden verwendet, um die Populationszusammensetzung zu analysieren und die Verwandtschaft zu bestimmen.
Ein Beispiel für eine Epistase sind die Variationen von rs7412 und rs429358 im APOE-Gen, wobei eine bestimmte Kombination von Varianten das Risiko für die Entwicklung der Alzheimer-Krankheit stark erhöht, während jede Variante einzeln nur einen geringen Beitrag zum Risiko leistet.
Laden Sie die stabile Version von Plink von der offiziellen
Website herunter .
BCFtools
BCFtools ist eine Reihe von Dienstprogrammen zur Bearbeitung genetischer Daten im VCF-Format und seinem binären Gegenstück BCF. Die Liste der möglichen Anwendungen des BCFtools-Pakets umfasst das Kommentieren, Filtern, Zusammenführen und Aufteilen von VCF / BCF-Dateien, das Finden ihrer Schnittpunkte, das Indizieren, das selektive Suchen, das Sortieren, das Zählen von Statistiken usw.
Gehen Sie zur Installation wie folgt vor:
git clone git://github.com/samtools/htslib.git git clone git://github.com/samtools/bcftools.git cd bcftools
Der Installationsvorgang wird
hier ausführlicher beschrieben.
KÖNIG
Das KING-Paket (Kinship-based INference for Gwas) wird in Populationsstudien verwendet, wenn mit Daten aus einer genomweiten Assoziationssuche gearbeitet wird, um Familienbeziehungen in den untersuchten Daten zu bestimmen. In dieser Aufgabe hilft KING dabei, den Verwandtschaftsgrad mehrerer Proben aus dem 1000-Genom-Projekt zu bestimmen.
Sie können es
hier herunterladen. Zur Behebung von Problemen finden Sie hier das KING-Handbuch.
Nahezu alle Fehler, die bei der Arbeit mit Werkzeugen auftreten können, sind in Stackoverflow oder seinem bioinformatischen Gegenstück - Biostars - beschrieben .
Verwendete Daten
Zur Orientierung verwenden wir die offenen Daten
aus dem 1000 Genomes-Projekt. Für die Analyse haben wir 10 Proben mit Informationen über die Genotypen von etwa 85 Millionen Variationen ausgewählt, die durch Analyse von NGS-Daten erhalten wurden, die mit der Version des Referenzgenoms GRCh37 übereinstimmen. Familienbeziehungen und Stichprobenpopulationen sind in Abbildung 1 dargestellt.
 Abbildung 1
Abbildung 1 Pedigree in VCF-Proben verwendet. Das Quadrat entspricht dem männlichen Geschlecht, der Kreis dem weiblichen. Gepunktete Linie bedeutet unbestimmte Verwandtschaft zweiter Ordnung.
Beachten Sie
Das VCF-Format ermöglicht es Ihnen, Informationen über das Feld einer Person als einzelne Zahl zu speichern, wenn diese Informationen während der Erstellung des VCF bekannt waren. Es sieht so aus: Das GT-Feld (Genotyp, Genotyp) für Datensätze aus dem X-Chromosom enthält einen numerischen Wert, der einem Allel entspricht, für Männer und zwei für Frauen. Wenn keine Informationen zum biologischen Feld der sequenzierten Probe vorliegen, enthält das GT-Feld standardmäßig zwei numerische Werte (in Abbildung 2 rot hervorgehoben).
In den in diesem Handbuch verwendeten VCF-Dateien ist das Y-Chromosom ausgeschlossen. Das Vorhandensein des Y-Chromosoms in der VCF-Datei bedeutet jedoch nicht immer, dass die sequenzierte Probe tatsächlich über dieses verfügt. Dies liegt an pseudoautosomalen Regionen (PARs), die für die X- und Y-Chromosomen identisch sind und sich an ihren Enden befinden.
Verschiedene Chromosomen haben normalerweise nicht lange identische (homologe) Regionen, jedoch besitzen X- und Y-Chromosomen solche Regionen, die zu Beginn (PAR1) und am Ende (PAR2) mehrere Millionen Basenpaare lang sind. Daher werden bei der Analyse von NGS-Daten bei Männern in den PAR-Regionen zwei Allele gefunden (eines für jedes Geschlechtschromosom), und bei Frauen können Genotypen in den PAR-Regionen des Y-Chromosoms auftreten, obwohl dies tatsächlich Genotypen aus ihrem X-Chromosom sind.
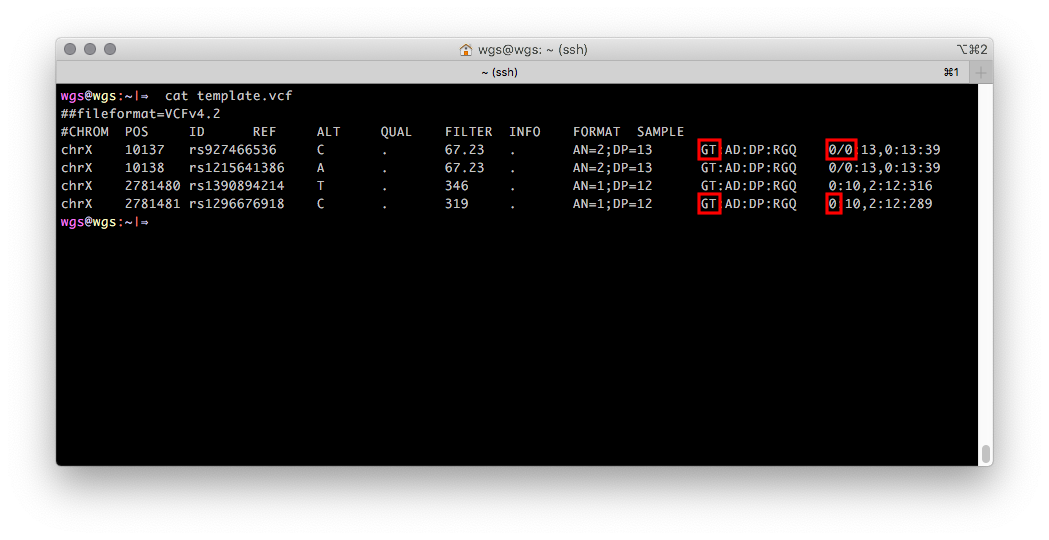 Abbildung 2
Abbildung 2 VCF-Datei mit Genotypen aus dem X-Chromosom eines Mannes aus der PAR1-Region (erste beiden Einträge) und der nicht-pseudo-autosomalen Region (letzte beiden Einträge).
Unterrichtseinheit
Das genetische Geschlecht ist ein Satz von Geschlechtschromosomen, die der Manifestation von primären und sekundären Geschlechtsmerkmalen eines männlichen oder weiblichen Typs entsprechen. Normalerweise haben Männer ein Chromosom X und ein Chromosom Y, während Frauen zwei Chromosomen X haben. Mit verschiedenen Störungen bei der Bildung von Keimzellen, Eiern und Spermien kann ein Kind mit einem ausgezeichneten Satz von Geschlechtschromosomen zur Welt kommen, was häufig zu Entwicklungsstörungen führt primäre und sekundäre sexuelle Merkmale.
Die beiden häufigsten chromosomalen sexuellen Anomalien sind das Turner-Syndrom (ein Satz von Chromosomen X0, dh nur ein X-Chromosom) und das Klinefelter-Syndrom (ein Satz von Chromosomen XXY).
Ein Allel ist ein oder mehrere Nukleotide, die sich an einer beliebigen Stelle im Genom befinden und eine Alternative haben. Das Konzept wird zur Beschreibung von Genotypen verwendet. Unterscheiden Sie zwischen Referenz-Allelen und Alternativen. Alle werden in der VCF-Datei in den Feldern REF und ALT gespeichert.
Bestimmen Sie das Geschlecht
Für Benutzer von Yandex.CloudAlle Daten zur Durchführung der manuellen und unabhängigen Aufgaben werden in Yandex.Cloud in der unten gezeigten Struktur gespeichert. Der
Tutorial Ordner enthält die VCF-Datei, die zur Vervollständigung des Handbuchs benötigt wird, den
Test Ordner für unabhängige Aufgaben. Der Ordner "
Technical " enthält zwei Dateien mit einer Liste der Identifikatoren genetischer Variationen: "
rsids_for_subsetting.txt im Handbuch verwendet und Aufgaben für die unabhängige Ausführung. "
external_interpretation_rsids.txt möglicherweise in Zukunft benötigt, wenn die genomweite Sequenzierung im Atlas zum Hochladen von Genotypisierungsdaten an Dienste von Drittanbietern erfolgt. Der Ordner
Tools enthält unter anderem zwei Skripte, die in den Aufgaben 2 und 3 verwendet werden.
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
Im Verzeichnis
/home auf der Yandex.Cloud-VM wird ein Ordner erstellt, dessen Name dem Benutzernamen entspricht, der bei der Erstellung der VM angegeben wurde. Kopieren Sie alles aus dem Verzeichnis
/home/ubuntu mit den folgenden Befehlen in Ihr Verzeichnis:
cd ~ cp -r /home/ubuntu/* ./
Für den RestWenn Sie an einem PC arbeiten, können Sie die für die erste Aufgabe erforderlichen Dateien über den
Link herunterladen. Das heruntergeladene Archiv unterstützt eine ähnliche Dateispeicherstruktur wie Yandex.Cloud:
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
Entpacken Sie das Archiv
atlas_wgs_contest.tar.gz mit dem Befehl
tar -xvzf atlas_wgs_contest.tar.gz VCF-Dateien für die Ausführung von Aufgaben in nicht archivierter Form belegen jeweils etwa 19 Gigabyte. Aus Platzgründen empfehlen wir daher, nur mit Archiven zu arbeiten. Alle oben aufgeführten Programme können bereits mit komprimierten VCF-Daten arbeiten. Darüber hinaus müssen Sie nichts tun.
Um das Geschlecht des Subjekts zu bestimmen, müssen Sie die Genotypen auf dem X-Chromosom untersuchen und die Regionen PAR1 und PAR2 am Anfang und Ende ausschließen. Dies sind die Intervalle der Positionen 60001–2699520 und 154931044–155260560 in der GRCh37-Version des Genoms. Wenn der Genotyp eine numerische Bezeichnung enthält, ist dies das männliche biologische Geschlecht, ansonsten das weibliche. Es ist zu beachten, dass die Benennung des Geschlechts in der VCF-Datei von der Verfügbarkeit von Informationen über das biologische Feld während der Erzeugung der VCF abhängt. Daher kann dieser Ansatz nicht immer verwendet werden.
Verwenden Sie für jedes Beispiel im Dataset den folgenden Befehl. Ersetzen Sie die Beispiel-ID nach dem Argument
-s :
(/Data/Tutotrial/CEI.1kg.2019.demo.vcf.gz):
Wenn Sie die Befehle ausführen, sehen Sie einen Teil des Inhalts der VCF-Datei für die angegebene Beispiel-ID. Der
-r chrX:2699521-154931043 in BCFtools beschränkt die Anzeige des Inhalts der Datei auf den Bereich von Chromosom X von Position 2699521 bis Position 154931043 (Nicht-PAR-Bereich in Abbildung 3). Diese Grenzen schließen pseudoautosomale Regionen aus, die in diesem Fall nicht erforderlich sind (PAR1 und PAR2). Bestimmen Sie anhand der numerischen Werte im GT-Feld das Geschlecht jeder Probe.
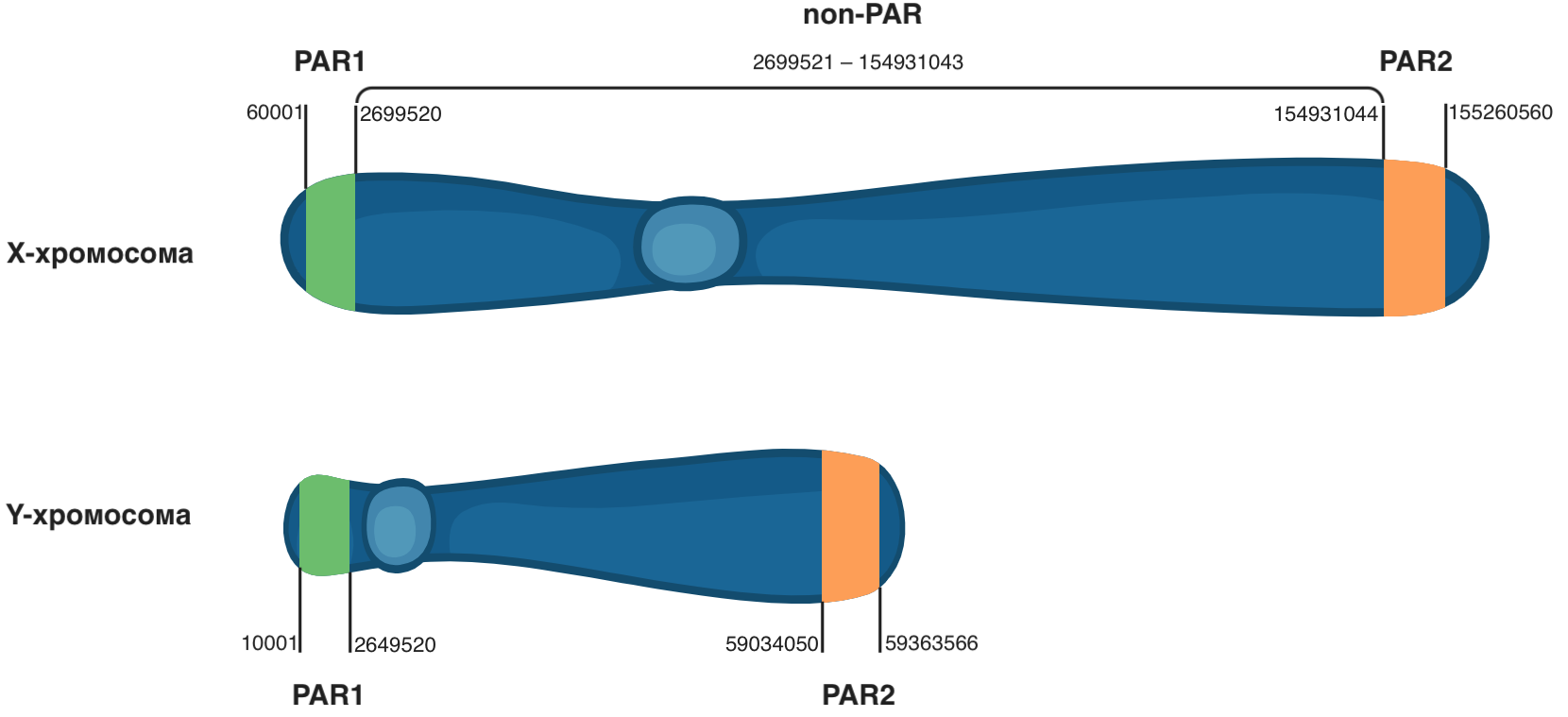 Abbildung 3
Abbildung 3 Die Position der pseudo-autosomalen Regionen PAR1 und PAR2 auf den Geschlechtschromosomen.
Sie können die Liste aller Beispiel-IDs in der VCF-Datei in Abbildung 1 oder in der letzten Zeile des Headers der VCF-Datei sehen. Sie werden nach dem Spaltennamen FORMAT aufgeführt:
Das wahre Geschlecht dieser Proben ist ebenfalls in Abbildung 1 dargestellt.
Wir bestimmen die Beziehung
Um die Beziehung zu bestimmen, müssen wir die genetischen Daten aller Proben paarweise vergleichen. Es ist schwierig, dies mit dem vollen Genom zu tun: In diesem Fall benötigt eine VCF-Datei zehn Gigabyte. Der von uns verwendete VCF nimmt nur etwa 2 Gigabyte in Anspruch, filtert ihn jedoch nach der Liste der genetischen Variationskennungen (rsIDs), die auf den Chips von Illumina genotypisiert sind: GSA v1, GSA v2, HumanOmniExpress v1.0, HumanOmniExpress v1.3, InfiniumExome v1. 1 und Infinium OmniExpressExome v1.4. Dies sind die beliebtesten Chips bei der kommerziellen Genotypisierung.
Wir haben eine Liste aller Identifikatoren genetischer Variationen dieser Chips in einer separaten Datei mit einer Liste von rsIDs zusammengestellt. Es enthält 1,4 Millionen Identifikatoren. Führen Sie den folgenden Befehl aus, um die VCF-Datei zu filtern:
bcftools view -O z -i 'ID=@rsids_for_subsetting.txt' CEI.1kg.2019.demo.vcf.gz > CEI.1kg.2019.demo.subset.vcf.gz
Jedes Mal, wenn Sie BCFtools und andere Pakete zum Arbeiten mit VCF-Dateien verwenden, wird der Verlauf vorheriger Befehle zum Header der Datei hinzugefügt. Unabhängig von der Methode zum Filtern der VCF-Datei und den zuvor ausgeführten Befehlen können Sie die Integrität und Identität des Hauptinhalts der VCF überprüfen, indem Sie die Hash-Summe berechnen:
Der Befehl
gunzip -c dekomprimiert die Datei mit der Ausgabe ihres Inhalts auf stdout, aus der die
# beginnenden Kopfzeilen der VCF-Datei weiter gelöscht werden (daher wird der Befehl
grep -v "^#" verwendet). Der Header wird entfernt, um nur die Integrität der genetischen Daten selbst zu vergleichen und nicht die Metadaten darüber, welche Tools und wann für die Arbeit mit dieser VCF-Datei verwendet wurden.
Wenn der Hash-Wert übereinstimmt, können Sie VCF in das interne Plink-Format konvertieren (standardmäßig besteht das Plink-Format aus drei Dateien mit den Erweiterungen bed, bim und fam). In diesen Dateien bleiben nur der Genotyp, das Chromosom, die Position und einige andere Daten übrig, und der Rest wird eliminiert. Mit diesem Format ist es viel einfacher, verschiedene Probleme zu bearbeiten und zu lösen, für die keine zusätzlichen Informationen von VCF erforderlich sind. Führen Sie beispielsweise GWAS durch.
Dieser Befehl erstellt drei Dateien im Ordner:
CEI.1kg.2019.demo.subset.bed
CEI.1kg.2019.demo.subset.bim
CEI.1kg.2019.demo.subset.famSie können für alle 10 Proben eine paarweise Verwandtschaft bestimmen. Wir verwenden den folgenden Befehl, um Plink-Dateien zu analysieren:
king -b CEI.1kg.2019.demo.subset.bed --kinship --prefix CEI.1kg.2019.demo.subset.kinship_analysis
Sehen Sie sich die Datei
CEI.1kg.2019.demo.subset.kinship_analysis.kin0 und achten Sie auf die Spalte Kinship, die die Verwandtschaftskoeffizienten für die in ID1 bzw. ID2 angegebenen Stichprobenpaare enthält.
Vergleichen Sie die in der Datei
CEI.1kg.2019.demo.subset.kinship_analysis.kin0 erhaltenen Koeffizienten für alle Stichprobenpaare mit dem in Abbildung 1 gezeigten Stammbaum (die gestrichelte Linie entspricht der Verwandtschaft zweiter Ordnung, es gibt jedoch keine genauen Verwandtschaftsdaten, d. H es kann Cousins, Tante / Neffe oder Onkel / Nichte geben). Versuchen Sie, Ihre eigenen Schlussfolgerungen darüber zu ziehen, welche Werte von Verwandtschaftskoeffizienten der Verwandtschaft erster und zweiter Ordnung entsprechen können.
HinweisAuszug aus der KING-Dokumentation: Verwandtschaftskoeffizienten> 0,354 entsprechen doppelten Stichproben oder identischen Zwillingen, 0,177 bis 0,354 Verwandtschaft erster Ordnung (Eltern-Kinder, Geschwister), 0,0884 bis 0,177 Verwandtschaft zweiter Ordnung (Cousins, Tanten) / Onkel-Neffen) und von 0,0442 bis 0,0884 - bis zur Verwandtschaft dritter Ordnung (Großeltern, Enkelkinder, zweite Cousins). Alles, was weniger als 0,0442 ist, ist schwer eindeutig zu interpretieren.
Die erste Aufgabe des Wettbewerbs
Data/Test/CEI.1kg.2019.test.vcf.gz anhand eines Testdatensatzes von 12 Stichproben
Data/Test/CEI.1kg.2019.test.vcf.gz ihren Stammbaum, der sich an den Ergebnissen der Geschlechtsbestimmung und der Verwandtschaftsanalyse orientiert. Proben, die nach den Ergebnissen der Analyse nicht mit jemandem verwandt sind, sollten in der Nähe notiert werden, ohne sie mit einer Linie mit anderen Proben zu verbinden. Der Stammbaum kann in einem ähnlichen Stil wie in Abbildung 1 verfasst werden, dies bleibt jedoch Ihrem Ermessen überlassen. Männer sind durch ein Quadrat gekennzeichnet, Frauen durch einen Kreis, die Ehe durch eine horizontale Linie, ein Kind durch eine vertikale Linie, mehrere Kinder durch eine horizontale Verzweigung einer vertikalen Linie (in Form des Buchstabens P). Lesen Sie hier mehr über diese Bezeichnungen.
Wie wir oben geschrieben haben, können Verwandtschaftskoeffizienten die Verwandtschaft der einen oder anderen Ordnung nicht eindeutig charakterisieren: Beim Vergleich der Paare Eltern-Kind und Bruder-Schwester (Verwandtschaft erster Ordnung) werden dieselben Verwandtschaftskoeffizienten erhalten. Wenn es nicht möglich ist, die Art der Beziehung festzustellen, geben Sie eine der möglichen an. Bitte beachten Sie, dass die Proben im Testdatensatz andere Bezeichner aufweisen als im Trainingsdatensatz.
Die Antworten
sollten bis zum 26. Dezember um 23:59 Uhr an
wgs@atlas.ru gesendet werden . Zwei weitere Aufgaben werden in Kürze veröffentlicht, und die endgültigen Ergebnisse für die Aufgaben werden am 28. Dezember veröffentlicht. Der Gewinner erhält den vollständigen Genomtest und die Plätze zwei und drei erhalten den Atlas-Gentest. Es wird auch Sonderpreise von
Yandex.Cloud geben . Ehemalige und aktuelle Mitarbeiter von Atlas nehmen nicht am Wettbewerb teil;)