Wir setzen den Aufgabenzyklus fort, in dem wir über den Umgang mit genetischen Daten sprechen. Die erste
Aufgabe „Finden Sie Geschlecht und Verwandtschaftsgrad heraus“ kann bereits gelöst und beantwortet werden. Heute veröffentlichen wir die zweite.
Der Hauptpreis ist das
komplette Genom .

Wir haben zuvor nützliche Informationen und Links geteilt, die für die Arbeit mit Bioinformatikdaten nützlich sein können. Wir empfehlen, dass Sie die vorherigen Artikel zuerst lesen, wenn Sie sie verpasst haben:
Was ist das vollständige Genom und warum wird es benötigt?Aufgabennummer 1. Finden Sie Geschlecht und Verwandtschaftsgrad heraus.Haftungsausschluss
Die Arbeit mit genetischen Daten wird auf Unix-Systemen (Linux, macOS) ausgeführt, da einige Befehle und Software unter Windows nicht verfügbar sind. Aus diesem Grund besteht eine der einfachsten Lösungen für Windows-Benutzer darin, eine virtuelle Linux-Maschine zu mieten.
Alle nachfolgend beschriebenen Vorgänge werden auf dem Befehlszeilenterminal ausgeführt. Bevor Sie beginnen, lernen Sie, wie Sie in einem Terminal mit Ihrem Betriebssystem arbeiten und Befehle verwenden, da einige davon möglicherweise das Betriebssystem und Ihre Daten beschädigen können.
Benötigte Software
Wir haben das
Image einer virtuellen Maschine (VM) mit der gesamten erforderlichen Software auf Yandex.Cloud gesammelt. Anweisungen zum Einrichten der VM und zum Installieren der Software finden Sie im vorherigen
Artikel mit Aufgabe Nr. 1.
Dieses Mal müssen Sie ein zweidimensionales Streudiagramm mit Daten erstellen, die mit der Analysemethode der Hauptkomponenten ermittelt wurden. Wir empfehlen, dieses Diagramm mit jeder für Sie geeigneten Software zu erstellen: Excel, Google Sheets, Python, R und andere.
Um die Aufgabe abzuschließen, benötigen Sie das Plink 1.9-Softwarepaket. Wenn Sie es noch nicht installiert haben (und Aufgabe Nr. 1 noch nicht abgeschlossen haben), lesen Sie den vorherigen Artikel. Es enthält Installationsanweisungen. Um am Neujahrswettbewerb 2019 teilnehmen zu können, müssen alle Aufgaben erledigt sein!
Beachten Sie
Die Hauptkomponentenanalyse (PCA) ist einer der lehrerlosen Algorithmen für maschinelles Lernen, wenn eine Maschine unabhängig nach Mustern in Daten sucht. In der Genetik ermöglicht PCA das Clustering von Proben nach Genotypisierungsdaten in einem N-dimensionalen Raum (normalerweise zweidimensional), wobei die wichtigsten Komponenten die Variabilität der genetischen Daten von Probe zu Probe am genauesten erklären.
Bei der Durchführung einer solchen Analyse bilden Proben einer Population normalerweise einen Cluster, dessen Größe und Glätte von der Ähnlichkeit der Proben innerhalb einer gegebenen Population abhängt. Der Algorithmus identifiziert wahrscheinlich Proben aus verschiedenen Populationen in verschiedenen Clustern. Und Proben von Populationen in der Nähe, die zur selben Superpopulation gehören, z. B. EAS - East Asian, wie in Abbildung 1, werden nahe beieinander oder sogar in sich überschneidenden Clustern identifiziert.
 Abbildung 1
Abbildung 1 Der Stammbaum der im VCF verwendeten Stichproben (das Quadrat entspricht dem männlichen Geschlecht, der Kreis dem weiblichen). Die gestrichelte Linie entspricht einer unbestimmten Beziehung zweiter Ordnung.
Eine ähnliche Analyse wird verwendet, um die Population durch Genotypisierung zu bestimmen. Hierzu wird ein Referenzdatensatz benötigt, der aus Proben mit bereits bekannter Herkunft besteht. Aus der Population lässt sich schließen, welche Gruppe bekannter Stichproben den untersuchten Daten am nächsten kommt.
Zur Vereinfachung besteht das Wesentliche der PCA-Analyse darin, dass paarweise Abstände zwischen Punkten im mehrdimensionalen Raum bekannt sind und diese Punkte in einem Raum kleinerer Dimension liegen müssen, damit sich die neuen paarweisen Abstände minimal von den ursprünglichen unterscheiden. Die Dimensionsreduktion vereinfacht die Datenanalyse, aber je mehr wir sie reduzieren, desto stärker unterscheiden sich die neuen Abstände zwischen Punkten vom Original. Daher besteht die Aufgabe der PCA-Analyse auch darin, einen Kompromiss zwischen Genauigkeit und einfacher Analyse zu finden. Alles ist wie im Leben.
Die einfachste Variante der PCA-Ausführung von genetischen Daten basiert auf der Identität einiger Allele, die in zwei Subtypen unterteilt werden können: IBS (Identität nach Status) und IBD (Identität nach Abstammung). IBS bedeutet die Identität bestimmter Allele bei zwei Personen, impliziert jedoch nicht notwendigerweise die Tatsache einer Beziehung zwischen ihnen. Im Gegensatz dazu spricht IBD von der Identität von Allelen aufgrund der Anwesenheit eines gemeinsamen Vorfahren und dementsprechend von Verwandtschaft.
IBD-Allele sind eindeutig IBS-Allele, während das Gegenteil nicht zutrifft. Es muss jedoch berücksichtigt werden, dass wir zu einem bestimmten Zeitpunkt von einem gemeinsamen Vorfahren abstammen, sodass einige Allele möglicherweise IBD sind. In der nachstehenden PCA-Analyse wird nur das IBS-Konzept verwendet, bei komplexeren Analysen werden jedoch Tests der statistischen Signifikanz, der phänotypischen Einschränkungen, der Clustergröße, des Alters und des Geschlechts der Person sowie zusätzliche Informationen zur Populationsstruktur berücksichtigt.
Je größer die Anzahl der verschiedenen Allele in zwei Proben ist, desto weniger ähneln sie sich und desto weiter sind sie voneinander entfernt. Der IBS-Wert für solche Proben ist niedrig. Aber für Eltern und ihre Kinder wird der IBS sehr hoch sein.
Wenn Sie die IBS-Werte für jedes Bildpaar in der Datenmenge kennen, können Sie eine PCA-Analyse durchführen, um festzustellen, wie sie gruppiert sind.
Der Atlas-Gentest verwendet einen viel ausgefeilteren Algorithmus zur Bestimmung der Populationsrepräsentation in Genotypisierungsdaten.
Verwendete Daten
Wir erinnern Sie daran, dass das Handbuch speziell ausgewählte offene Daten aus dem
1000 Genomes- Projekt verwendet. Für die Analyse wurden 10 Proben mit Genotypinformationen von ~ 85 Millionen Variationen ausgewählt, die durch Analyse von NGS-Daten erhalten wurden, die mit der Version des GRCh37-Genoms übereinstimmen. Familienbeziehungen und Populationen dieser Stichproben sind in Abbildung 1 dargestellt.
Aufbau von Bevölkerungsclustern
Verwenden Sie die drei Dateien im Plink-Format, die Sie zuvor in Aufgabe 1 erhalten haben:
CEI.1kg.2019.demo.subset.bed CEI.1kg.2019.demo.subset.bim CEI.1kg.2019.demo.subset.fam
Bestimmen Sie den paarweisen Abstand zwischen allen 10 Proben im Trainingsdatensatz und zeichnen Sie eine PCA basierend auf IBS (Identity by State). Dies kann wie folgt erfolgen:
Der
—genome Parameter ist nur für die paarweise Berechnung von IBS / IBD zwischen allen Proben im Datensatz verantwortlich. Der Parameter "
—read-genome " ist die zuvor erhaltene paarweise Distanzmatrix, und die Parameter "
—cluster —mds-plot 10 sind für die PCA-Analyse und die Ausgabe ihrer Ergebnisse an die Tabelle der 10 ersten Hauptkomponenten verantwortlich. Tatsächlich sind dies die Koordinaten jeder Probe im 10-dimensionalen Raum.
Mit dem letzten Befehl werden 4 Dateien im Ordner erstellt:
CEI.1kg.2019.demo.subset.clustering.cluster1 CEI.1kg.2019.demo.subset.clustering.cluster2 CEI.1kg.2019.demo.subset.clustering.cluster3 CEI.1kg.2019.demo.subset.clustering.mds
Wir benötigen die letzten beiden Dateien aus der Liste.
Abbildung 2 zeigt, wie die im MDS-Trainingsdatensatz empfangene Datei aussieht. Die Felder FID (Family ID) und IID (Individual ID) entsprechen den Kennungen der Familie und der einzelnen Proben. Die Felder C1 - C10 enthalten die Werte jeder der zehn Hauptkomponenten für jede Probe, wobei Komponente C1 die Variabilität der genetischen Daten der analysierten Proben maximal erklärt und C10 minimal.
 Abbildung 2
Abbildung 2 MDS-Datei mit Werten von 10 Hauptkomponenten für jede Probe.
Bei der Erstellung eines Streudiagramms mit zwei Komponenten (im zweidimensionalen Raum) kann man Cluster erkennen, die der Population der Probe entsprechen. 3 zeigt Streudiagramme für die Paare der Hauptkomponenten C1xC2, C2xC3 und C1xC3. Beim Vergleich der erhaltenen Cluster mit der Referenzpopulationszugehörigkeit (Abbildung 1) zeigt das Paar der ersten beiden Komponenten C1 - C2 die höchste Genauigkeit (100%) und trennt alle Proben korrekt gemäß ihrer im 1000-Genom-Projekt angegebenen Populationszugehörigkeit. Aufgrund der möglichen Überlappung oder Trennung realer Cluster ist es jedoch immer sinnvoll, die Ergebnisse für mehrere Komponentenpaare zu vergleichen.
 Abbildung 3
Abbildung 3 Streudiagramme von Probenorten für Paare von Hauptkomponenten; Die Position der Marker wurde leicht geändert, um ein Überlappen zu verhindern.
Das Erstellen von 3D-Diagrammen unter Verwendung der ersten drei Hauptkomponenten kann ebenfalls hilfreich sein, um die Clusterbildung zu bestimmen, jedoch nicht immer. Wenn Sie beispielsweise ein solches Diagramm für die Daten in Abbildung 3 erstellen, können Sie vier Cluster identifizieren, in denen Proben aus der PUR- und KHV-Population nach Populationen gruppiert sind, und Proben aus der CDX-Population in zwei Cluster unterteilt sind (Abbildung 4). Dies macht sich auch in Abbildung 3 in den Koordinaten C2xC3 und C1xC3 bemerkbar.
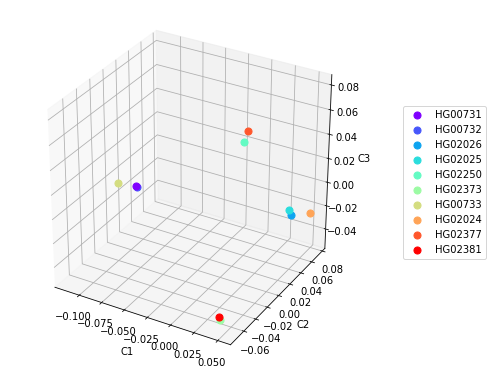 Abbildung 4
Abbildung 4 Streudiagramme für die drei Hauptkomponenten.
Solche widersprüchlichen Analyseergebnisse lassen sich durch eine geringe Anzahl von Stichproben erklären, da die Werte der Hauptkomponenten jeder Stichprobe für unterschiedliche Datensätze in Bezug auf Größe und Zusammensetzung unterschiedlich sind und sich das Ergebnis der Clusterbildung ändern kann, wenn zusätzliche Stichproben aus unterschiedlichen Populationen einbezogen werden. Möglicherweise sind auch Fehler beim Erstellen eines Datensatzes und beim Bereitstellen von Referenzdaten zur Population von Proben möglich. Im 1000-Genom-Projekt ist die Wahrscheinlichkeit einer solchen Situation jedoch recht gering.
In einer MDS-Datei werden keine Tabulatoren oder Kommas als Trennzeichen verwendet. Passen Sie daher das Format an, um die Eingabe zu vereinfachen. Verwenden Sie
tab oder
csv als zweites Argument:
Das Team erstellt die Datei
CEI.1kg.2019.demo.subset.clustering.mds.tab , die Sie herunterladen und Streudiagramme erstellen
CEI.1kg.2019.demo.subset.clustering.mds.tab , die den in Abbildung 3 gezeigten ähnlich sind. Vergleichen Sie die Ergebnisse, sie sollten mit den oben angegebenen identisch sein.
Erstellen eines Clusterbaums
Sie können das Clustering von Samples auch mithilfe eines Binärbaums auswerten, der die Clustering-Informationen zu Samples in diskreter Form darstellt. Informationen zu diesem Baum sind in der Datei
CEI.1kg.2019.demo.subset.clustering.cluster3 enthalten (Abbildung 5).
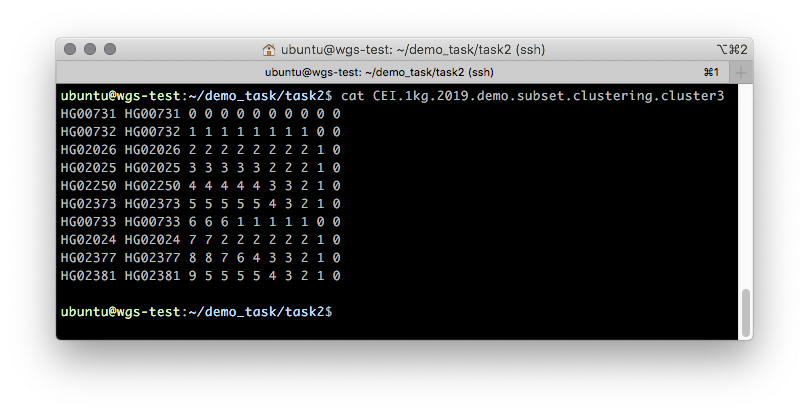 Abbildung 5
Abbildung 5 Der ungefähre Inhalt einer
.cluster3 Datei, die den Prozess der schrittweisen Clusterbildung von Stichproben von 1 Cluster zu N beschreibt, wobei N die Anzahl der Stichproben ist.
Die ersten beiden Spalten dieser Datei enthalten die FID und IID. Die Clusterzugehörigkeit wird von allen anderen beschrieben. Diese Datei sollte in Spalten in Schritten von einer Spalte von rechts nach links gelesen werden: Zunächst gehören alle Stichproben zu einem Cluster „0“ - der äußersten rechten Spalte. Bei Aufteilung in zwei Cluster (im zweiten Schritt in der zweiten Spalte) werden zwei Cluster angezeigt: "0" und "1", wobei der Cluster "0" die Beispiele HG00731, HG00732 und HG00733 enthält und der Cluster "1" den Rest enthält. Eine Darstellung einer solchen Partition ist in Abbildung 6 dargestellt.
Aus dem Baum kann geschlossen werden, dass die Stichproben zur Population gehören (Abbildung 1). Darüber hinaus können wir durch die Konstruktion dieses Baums die Nähe einzelner Populationen feststellen, nämlich das Auftreten der CDX- und KHV-Populationen in einer EAS-Superpopulation (bereits im ersten Schritt der Aufteilung der Superpopulationen werden EAS und AMR in zwei bestehende Zweige aufgeteilt). Die Erstellung eines Clusterbaums kann auch dazu beitragen, die mehrdeutigen Ergebnisse der Visualisierung von Proben auf den Hauptkomponenten zu korrigieren.
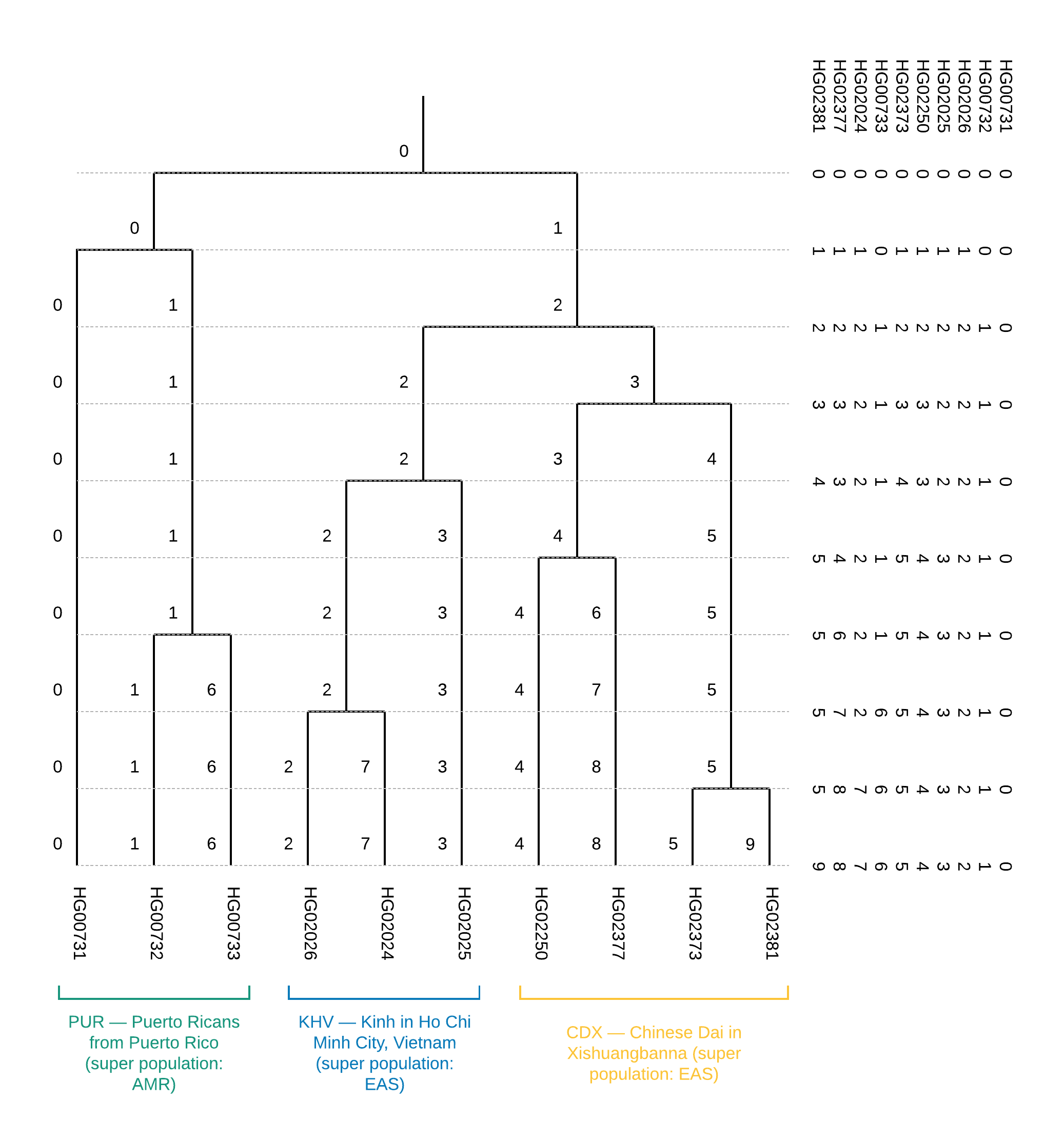 Abbildung 6
Abbildung 6 Binärer Clusterbaum für einen Trainingsdatensatz von 10 Stichproben: Auf der rechten Seite befindet sich der Inhalt der Datei
CEI.1kg.2019.demo.subset.clustering.cluster3 (von rechts nach links in der Datei, identisch von oben nach unten in der Abbildung).
Die zweite Aufgabe des Wettbewerbs
Verwenden Sie den Testdatensatz aus 12 Beispielen von
Data/Test/CEI.1kg.2019.test.vcf.gz und das obige Beispiel (Abbildung 5), um aus der erhaltenen
.cluster3 Datei einen binären Clusterbaum zu
.cluster3 und an die Lösung anzuhängen. Analysieren Sie den resultierenden Baum und ziehen Sie Schlussfolgerungen über die Anzahl der im Testdatensatz dargestellten Superpopulationen.
Bestimmen Sie die Populationshäufung von 12 Proben aus dem Testdatensatz, indem Sie die Hauptkomponenten C1, C2 und C3 unter Berücksichtigung des konstruierten Baums analysieren und dies in der in Aufgabe Nr. 1 erstellten Ahnentafel angeben, indem Sie einzelne Populationsblöcke einschränken (ähnlich wie in Abbildung 1). Proben, die in Problem Nr. 1 keine Verwandtschaft aufwiesen, müssen auf die gleiche Weise in den im Diagramm erhaltenen Blöcken platziert werden, ohne sie mit Linien mit anderen Proben zu verbinden. Vergessen Sie nicht, die von Ihnen erstellten Streudiagramme beizufügen.
Die Antworten
sollten bis zum 26. Dezember um 23:59 Uhr an
wgs@atlas.ru gesendet werden . Eine weitere Aufgabe wird in Kürze veröffentlicht, und die endgültigen Ergebnisse für die Aufgaben werden am 28. Dezember veröffentlicht. Der Gewinner erhält den vollständigen Genomtest und die Plätze zwei und drei erhalten den Atlas-Gentest. Es wird auch Sonderpreise von
Yandex.Cloud geben . Ehemalige und aktuelle Mitarbeiter von Atlas nehmen nicht am Wettbewerb teil;)