Heute veröffentlichen wir die letzte Aufgabe aus dem Zyklus, in dem wir erklären, wie man mit genetischen Daten arbeitet.
Die erste und
zweite Aufgabe sind bereits veröffentlicht: Sie können gelöst und beantwortet werden. Wir warnen Sie, dass diese Aufgabe länger dauert als der Rest.
Der Hauptpreis ist das
komplette Genom .

Wir haben zuvor nützliche Informationen und Links geteilt, die für die Arbeit mit Bioinformatikdaten nützlich sein können. Wir empfehlen, dass Sie die vorherigen Artikel zuerst lesen, wenn Sie sie verpasst haben:
Was ist das vollständige Genom und warum wird es benötigt?Aufgabennummer 1. Finden Sie Geschlecht und Verwandtschaftsgrad heraus.Aufgabennummer 2. Bestimmung der BevölkerungsstrukturHaftungsausschlussDie Arbeit mit genetischen Daten wird auf Unix-Systemen (Linux, macOS) ausgeführt, da einige Befehle und Software unter Windows nicht verfügbar sind. Aus diesem Grund besteht eine der einfachsten Lösungen für Windows-Benutzer darin, eine virtuelle Linux-Maschine zu mieten.
Alle nachfolgend beschriebenen Vorgänge werden auf dem Befehlszeilenterminal ausgeführt. Bevor Sie beginnen, lernen Sie, wie Sie in einem Terminal mit Ihrem Betriebssystem arbeiten und Befehle verwenden, da einige davon möglicherweise das Betriebssystem und Ihre Daten beschädigen können.
Benötigte Software
Wir haben das
Image einer virtuellen Maschine (VM) mit der gesamten erforderlichen Software auf Yandex.Cloud gesammelt. Anweisungen zum Einrichten einer VM und zum Installieren von Software finden Sie im
Artikel mit der ersten Aufgabe. Es gibt auch eine Anleitung, wie Sie das Gerät so einrichten, dass es bis zum 31. Dezember 2019 kostenlos verwendet werden kann.
In dieser Aufgabe müssen Sie die Genotypisierungsdaten vom VCF-Format in das 23andMe-Format konvertieren, die empfangenen Dateien zum Promethease-Service hochladen und sich mit dem Inhalt des Berichts für jede Probe vertraut machen.
Das 23andMe-Format ist ein Textformat zum Speichern von Genotypisierungsdaten und enthält 4 durch Tabulatoren getrennte Felder. Das erste Feld enthält den Variationsbezeichner (z. B. rsID), das zweite enthält das Chromosom (gültige Werte für dieses Feld sind 1-22, X, Y und MT), das dritte enthält die Position auf dem Chromosom, das vierte enthält den Genotyp (diploid in Gegenwart von zwei homologen Chromosomen, haploide in anderen Fälle). Dieses Format wird von vielen Dolmetschdiensten unterstützt, daher werden wir in der Aufgabe damit arbeiten.
Um die Aufgabe abzuschließen, benötigen Sie das BCFtools-Softwarepaket. Wenn Sie es noch nicht installiert haben, lesen Sie den
Artikel mit der ersten Aufgabe. Es enthält Installationsanweisungen. Wir erinnern Sie daran, dass zur Teilnahme am Neujahrswettbewerb 2019 alle Aufgaben erledigt sein müssen.
Zusätzlich zu BCFtools müssen Sie die Datei "
create_23andme.sh erstellen - ein Bash-Skript, mit dem Daten im 23andMe-Format generiert werden. Diese Datei befindet sich im Verzeichnis
/Technical auf Yandex.Cloud sowie im Archiv zum Herunterladen, das über den Link im
Artikel verfügbar ist.
Beachten Sie
Es gibt viele Dienste, die Genotypisierungsdaten analysieren: MyHeritage, Promethease, FamilyTreeDNA, DNA.LAND, GEDmatch. Sie bieten den Download von Genotypisierungsdaten in verschiedenen Formaten, häufig spezifisch für einen bestimmten Genotypisierungsanbieter (Ancestry, 23andMe, MyHeritage, FamilyTreeDNA, GenesForGood und andere). Promethease ist dem Datenformat am treuesten: Sie können sowohl VCF- als auch 23andMe-Dateien in diesen Dienst herunterladen.
Es gibt verschiedene Kompatibilitätsprobleme zwischen Formaten und Diensten:
- Verschiedene Unternehmen verwenden unterschiedliche Versionen des Genoms, um genetische Variationen abzubilden.Dieses Problem wird durch den sogenannten Liftover gelöst, wenn die Positionen genetischer Variationen in den Quelldaten durch die entsprechenden Positionen in einer anderen Version des Genoms ersetzt werden. Beispielsweise stellt Atlas Genotypisierungsdaten für die Version des GRCh38-Genoms bereit und GEDmatch empfängt Daten für die vorherige Version des GRCh37-Genoms. Die Umrechnung der Koordinaten genetischer Variationen von GRCh38 nach GRCh37 wird als Elevator bezeichnet.
- Verwendung eindeutiger Identifikatoren für andere genetische Variationen als rsIDs. Solche Inkompatibilitäten werden behoben, indem solche Einträge aus der Datei ausgeschlossen oder durch Zuweisen einer rsID mit Anmerkungen versehen werden. Der zweite ist nicht immer möglich.
- Die Dienste verwenden einen festen Satz genetischer Variationen. Manchmal führt eine Nichtübereinstimmung von mindestens einem Teil der heruntergeladenen Daten zu einem Ladefehler. Dieses Problem ist beispielsweise für MyHeritage relevant. Es kann gelöst werden, indem eine Reihe von Identifikatoren genetischer Variationen hervorgehoben werden, die keinen Ladefehler verursachen.
Verwendete Daten
Wir erinnern Sie daran, dass dieses Handbuch speziell ausgewählte offene Daten aus dem
1000 Genomes- Projekt verwendet. Für die Analyse wurden 10 Proben mit Genotypinformationen von ca. 85 Millionen Variationen ausgewählt, die durch Analyse von NGS-Daten erhalten wurden, die mit der Version des GRCh37-Genoms übereinstimmen. Familienbeziehungen und Populationen dieser Stichproben sind in Abbildung 1 dargestellt.
 Abbildung 1
Abbildung 1 Der Stammbaum der im VCF verwendeten Stichproben (das Quadrat entspricht dem männlichen Geschlecht, der Kreis dem weiblichen). Die gestrichelte Linie entspricht einer unbestimmten Beziehung zweiter Ordnung.
VCF-Konvertierung
Nachstehend finden Sie Anweisungen zum Konvertieren einer VCF-Datei und zum Hochladen der empfangenen Daten zum kürzlich kostenlosen Promethease-Dienst. Wir empfehlen Ihnen, sich mit dem Promethease-Bericht vertraut zu machen, den Sie für eine der Proben erhalten haben. Verwenden Sie die VCF-Datei, die nach der Liste der in
Aufgabe Nr. 1 erhaltenen Variationen gefiltert ist.
Mit
bcftools query Befehl
bcftools query können Sie alle verfügbaren Informationen in einem vom Benutzer nach dem Flag
-f angegebenen Format aus einer VCF-Datei extrahieren. Das Flag
-s gibt die Kennung der Probe (
HG00731 ) an, für die Daten extrahiert werden sollen. Mit dem Flag -e werden Ausschlusskriterien angegeben, in diesem Fall
'%ID=="."' Schließt Einträge ohne rsID aus. Die Ausgabe der
bcftools query wird an das Skript
create_23andme.sh , das die Daten in das TSV-Format mit 4 Spalten (rsID, Chromosom, Position, Genotyp) konvertiert und in eine Datei schreibt. Sie können das Skript
create_23andme.sh herunterladen und für sich selbst speichern, um mit Ihren eigenen Sequenzierungsdaten für das gesamte Genom zu arbeiten.
Das Skript
create_23andme.sh verwendet die aus der VCF-Datei extrahierten
create_23andme.sh , um den Typ der genetischen Variation (Single-Nucleotide-Variation von SNV, Insertion von INS oder Deletion von DEL) zu bestimmen, und schreibt den Bezeichner rsID, Chromosom, Position und Allele in
stdout entsprechend dem spezifischen Variationstyp (A, G, T) und C sind gültige Allele für den Typ SNV, I und D sind gültige Allelbezeichnungen für die Typen INS und DEL.
Beachten Sie, dass der Konvertierungsprozess viel Zeit in Anspruch nimmt: ca. 4 Stunden pro Datei für ein Sample mit ca. 1 Million Variationen. Parallelität BCFtools wird nicht unterstützt.
Gehen Sie auf
promethease.com und registrieren Sie sich. Klicken Sie auf die Schaltfläche Rohdaten hochladen (Abbildung 2) und laden Sie die Datei
HG00731.subset.23andme.txt . Nachdem der Download abgeschlossen ist, klicken Sie auf die Schaltfläche Kostenloser Bericht erstellen und geben Sie den gewünschten Namen des Berichts ein, der gemäß Ihren Daten erstellt wird. Nach der Erstellung des Berichts erhalten Sie eine Benachrichtigung per E-Mail und können sich mit dem Inhalt des Berichts vertraut machen. Finden Sie in den Berichten für jede Probe die durch das Promethease-Interpretationssystem bestimmte Blutgruppe im AB0 / Rh-System (Rh-Rh-Faktor). Überprüfen Sie Ihre Ergebnisse auf Übereinstimmung mit Tabelle 1.
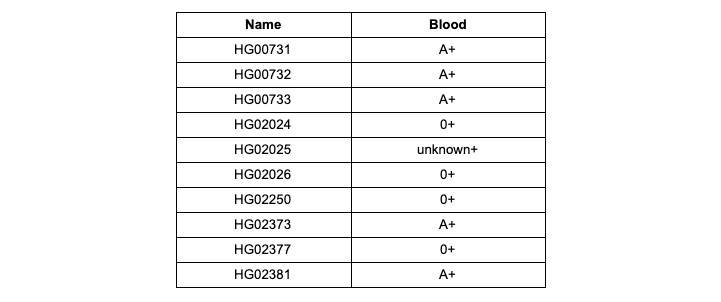 Tabelle 1
Tabelle 1 . Blutgruppen und Rh-Faktor aus einer Promethease-Analyse von Proben aus einem Demo-Datensatz
Der Atlas verwendet Schwellenwerte, die sich von Promethease unterscheiden, um ein bestimmtes Attribut in die Interpretation nach Evidenzgrad einzubeziehen. Der Evidenzgrad bezieht sich auf die Gesamtheit der Ergebnisse statistischer Tests und der Kriterien für die Signifikanz jeder Beziehung, die zwischen genetischer Variation und jeglichen Merkmalen des menschlichen Körpers beobachtet wird. Viele der Merkmale, die im Promethease-Bericht zu finden sind, weisen nur einen geringen und / oder einen hohen Evidenzgrad für eine begrenzte Anzahl von Populationen auf, beispielsweise nur für Vertreter der asiatischen Bevölkerung.
HinweisEmpirisch haben wir eine Liste genetischer Variationen auf der Basis des
Infinium Global Screening Array v2.0- Chips installiert, die auf MyHeritage hochgeladen werden können. Diese Liste (
external_interpretation_rsids.txt ) wird in einer separaten Datei im Verzeichnis
/Technical gespeichert und kann zum Filtern von VCF mit anschließender Konvertierung analog zu den obigen Anweisungen verwendet werden. Sie können diese Datei auch verwenden, um Genotypisierungsdaten von einem Chip zu filtern, damit Sie sie auf MyHeritage hochladen können. Wenn Sie bereits über den Atlas-Gentest verfügen, können Sie die Genotypisierungsdaten im Format von Ihrem persönlichen Konto hochladen und sie gemäß der vorgeschlagenen Variationsliste filtern - der ersten Spalte in den Daten, die von Ihrem persönlichen Konto hochgeladen wurden.
Beachten Sie, dass die in diesem Handbuch verwendeten Dateien immer ein ausgefülltes ALT-Feld (alternatives Allel) enthalten. Auf diese Weise können Sie nachvollziehen, zu welchem Typ jede Variation gehört (INS, DEL, SNV) und einen Eintrag im 23andMe-Format erstellen. Die genomweiten Sequenzierungsdaten im Atlas enthalten das gefüllte ALT-Allel nur an den Stellen, an denen dieses Allel gefunden wurde, da sonst die Informationen zum Füllen des ALT-Feldes beim Erstellen einer VCF-Datei einfach nicht vorhanden sind. Die Datenausgabe an homozygoten Referenzorten (Positionen im Genom, an denen das Referenzallel nicht gefunden wurde) ist notwendig, da nicht nur die nachgewiesenen Variationen der Nukleotidsequenz einen klinischen Effekt haben, sondern auch deren Abwesenheit.
Die Abwesenheit des ALT-Allels an solchen Positionen des Genoms erlaubt es uns nicht, die Art der genetischen Variation zu bestimmen, für die nur das Referenz- (REF) Allel gefunden wurde. Das Aufzeichnen von Genotypen für solche Fälle wird durch die Notwendigkeit erschwert, eine Informationsquelle über mögliche Allele für diese Variation zu verwenden, und wird in diesem Handbuch nicht behandelt. Wenn Sie dieses Handbuch und das Skript
create_23andme.sh möglicherweise verwenden, um eine nach einer genomweiten Sequenzierung in Atlas erhaltene VCF-Datei zu konvertieren, enthält die konvertierte Datei keine homozygoten Referenzgenotypen, da das Skript
create_23andme.sh solche Datensätze explizit filtert, um Fehler beim Erstellen von Datensätzen zu beseitigen für Einfügungen und Löschungen.
Damit das Skript
create_23andme.sh weiterhin homozygote Referenzgenotypen erzeugt, müssen Sie den Inhalt der Zeilen 25–28 ersetzen
... if [ "$ALT" == "." ] || [[ "$ALT" == *"*"* ]] then continue fi ...
auf
... if [[ "$ALT" == *"*"* ]] then continue fi if [ "$ALT" == "." ] then echo -e "$RSID\t$CHR\t$POS\t$REF$REF" fi ...
Durch diese Substitution können
stdout mit homozygoten Referenzgenotypen angezeigt werden. Es ist zu beachten, dass solche Einträge für Einfügungen und Löschungen falsch sind, da die gültigen Allele in dem für Einfügungen und Löschungen verwendeten Format I und D sind und das Skript die Allele A, G, T oder C. verwendet, um Daten für korrekt auszugeben Insertionen und Deletionen müssen im Voraus bekannt sein, welche Art von Variation für eine bestimmte Position des Genoms charakteristisch ist, in der das ALT-Allel nicht nachgewiesen wurde. Diese Informationen erhalten Sie, indem Sie das ALT-Allel analysieren, sofern es verfügbar ist (bereits in
create_23andme.sh implementiert) oder eine externe Datenbank verwenden, z. B. dbSNP (nicht in
create_23andme.sh ).
Um einen Promethease-Bericht über eine vollständige VCF-Datei mit vollständiger Genomsequenzierung in Atlas zu erhalten, können Sie die VCF-Datei selbst in Promethease laden. Beachten Sie jedoch, dass die Größe der komprimierten Atlas-VCF-Datei etwa 8 Gigabyte beträgt, während Sie mit Promethease maximal Dateien hochladen können 4 Gigabyte. Eine Beschreibung der Lösungen für dieses Problem finden Sie
hier . Eine andere Lösung besteht darin, die VCF-Datei in mehrere Teile aufzuteilen (jeweils weniger als 4 Gigabyte) und diese als zusätzliche Datei im Promethease-Menü zum Herunterladen von Daten zu laden.
Die dritte Aufgabe des Wettbewerbs
Laden Sie die konvertierten Daten jeder der 12 Proben des Testdatensatzes, die Sie gemäß der Liste der Variationen in der ersten Aufgabe gefiltert haben, in Promethease herunter und erstellen Sie eine Entsprechungstabelle für die vom Promethease-Interpretationssystem festgelegte Probenidentifikation - Blutgruppe AB0 / Rh (Rh-Faktor). Blutgruppen, die probabilistisch identifiziert und mit dem Präfix "prob" im Promethease-Bericht aufgezeichnet wurden, ohne Präfix schreiben. Notieren Sie undefinierte Werte als unbekannt (Rhesusfaktor für unbekannte Blutgruppen muss noch geschrieben werden, falls definiert). Ein Beispiel ist in Tabelle 1 dargestellt.
Das Konvertieren von VCF in das oben in der vorgeschlagenen Implementierung verwendete Format ist stark vereinfacht, erfordert jedoch einen erheblichen Zeitaufwand. Zur Optimierung können Sie ein Skript mit einer Schleife schreiben, die diese Daten automatisch generiert und über eine Reihe von Bezeichnern iteriert. Es ist möglich, mehrere solcher Skripte zu erstellen und jeweils verschiedene Sätze von Beispiel-IDs für die parallele Ausführung zu übertragen. Die Anzahl der parallel ausgeführten Skripte sollte jedoch die CPU-Anzahl Ihres Computers / Ihrer virtuellen Maschine nicht überschreiten. Eine gute Beschreibung zum Erstellen solcher Schleifen finden Sie
hier . Wenn Sie an Yandex.Cloud arbeiten, können Sie bei Bedarf eine weitere virtuelle Maschine mit einer großen Anzahl virtueller CPUs erstellen, wodurch sich der Zeitaufwand für die Ausführung einer Aufgabe proportional verringert.
Dies ist die letzte Aufgabe unseres Zyklus. Die Antworten
sollten bis zum 26. Dezember um 23:59 Uhr an
wgs@atlas.ru gesendet werden . Wir werden die richtigen Antworten und die Namen der Gewinner am 28. Dezember veröffentlichen. Der Gewinner erhält den vollständigen Genomtest und die Plätze zwei und drei erhalten den Atlas-Gentest. Es wird auch Sonderpreise von
Yandex.Cloud geben . Ehemalige und aktuelle Mitarbeiter von Atlas nehmen nicht am Wettbewerb teil;)