Cuando el morfólogo británico George Jackson Mywart [St. George Jackson Mivart] publicó en 1865 uno de los primeros árboles evolutivos, carecía de material de apoyo. Él construyó un árbol, un
mapa ramificado de varias especies de primates, utilizando un análisis detallado de las espinas de los animales. El segundo árbol, basado en una comparación de extremidades animales,
mostró otros lazos familiares entre primates, destacando el problema de la biología evolutiva que existe hasta el día de hoy.
Casi 150 años después, los científicos adquirieron montañas de datos para construir los llamados
árboles filogenéticos , una versión moderna de la estructura creada por Mivart. Los avances en la tecnología de decodificación de ADN y la
bioinformática le permiten comparar las secuencias de cientos de genes, y a veces genomas completos, de diferentes especies, y crear el árbol de la vida con más detalle que nunca.
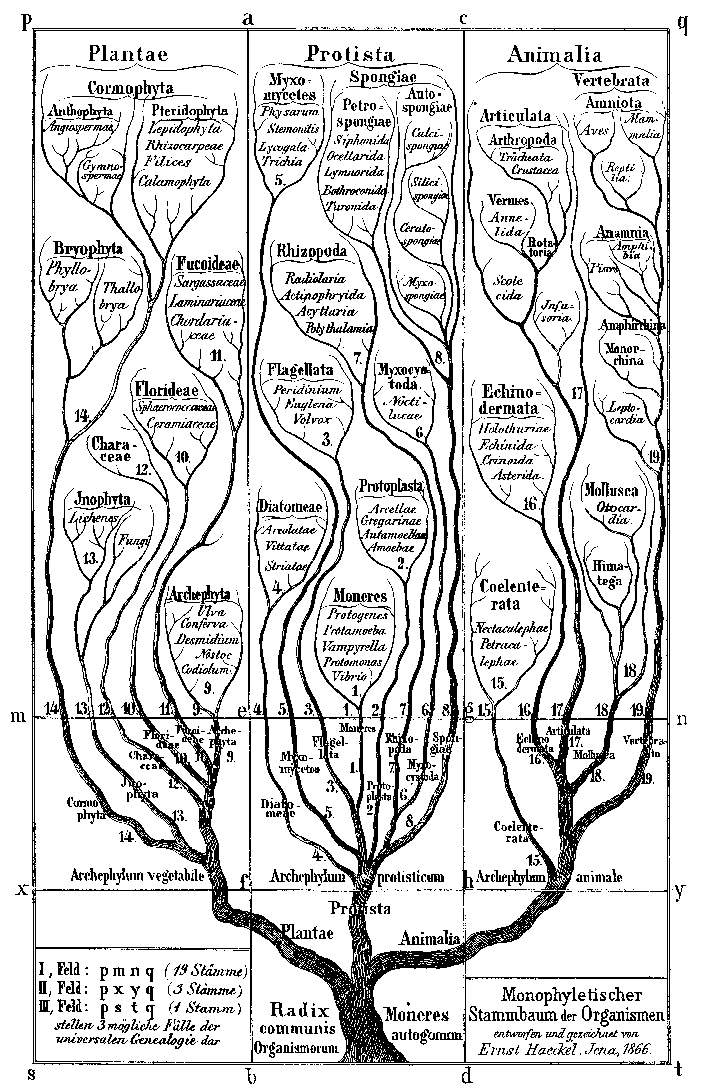 El árbol histórico de la vida de 1866 describe los reinos de plantas, animales y unicelulares.
El árbol histórico de la vida de 1866 describe los reinos de plantas, animales y unicelulares.Pero aunque la abundancia de datos ayudó a resolver algunos de los conflictos que surgieron en diferentes partes del árbol evolutivo, también trajo nuevas dificultades. La versión actual del árbol de la vida se parece más a una controvertida página de Wikipedia que a un libro publicado: hay debates en curso sobre algunas ramas. De la misma manera que la columna vertebral y las extremidades dieron lugar a la aparición de mapas conflictivos de la evolución de los primates, los científicos ahora saben que diferentes genes en el mismo organismo pueden contar historias diferentes.
Según un nuevo estudio, basado en parte en el estudio de la levadura, el cuadro controvertido dibujado por genes individuales es aún más contradictorio de lo esperado. "Se afirma que cada uno de los 1070 genes está involucrado en algún tipo de conflicto", dice Michael
Donoghue , un biólogo evolutivo de Yale que no está asociado con el estudio. "Estamos tratando de descubrir las relaciones filogenéticas de 1,8 millones de especies, y no podemos clasificar veinte tipos de levadura nosotros mismos", dice.
Para resolver la paradoja, los investigadores desarrollaron un algoritmo basado en la teoría de la información para medir el nivel de confianza en la corrección de las partes individuales del árbol. Esperan que el nuevo enfoque ayude a aclarar los períodos evolutivos que tienen los datos más interesantes y útiles y más conflictivos, por ejemplo, la
explosión cámbrica , la rápida diversificación de la vida animal que ocurrió hace 540 millones de años.
"Históricamente, los episodios más interesantes están relacionados con áreas que atrajeron la atención y causaron controversia", como el origen de animales, vertebrados y plantas con flores, dice
Antonis Rokas , biólogo de la Universidad de Vanderbilt, quien dirigió el nuevo estudio.
Según los resultados del nuevo algoritmo, los científicos pueden seleccionar solo los genes más informativos para construir árboles filogenéticos. Tal enfoque puede hacer que el proceso sea más preciso y eficiente. "Creo que ayudará a acelerar la reconstrucción del árbol de la vida", dijo Khidir Hilu, biólogo del Instituto de Tecnología de Virginia.
Ladrillos de la vida
La base de los árboles filogenéticos se crea a través de la agrupación de especies de acuerdo con su grado de parentesco. Si comparamos el ADN de humanos, chimpancés y peces, queda claro que los humanos y los chimpancés están más cerca el uno del otro que los peces.
Érase una vez, los investigadores utilizaron uno o más genes para comparar organismos. Pero en la última década ha habido una explosión de datos filogenéticos, llenando muy rápidamente las bases necesarias para crear estos árboles. El análisis llenó varios de los puntos blancos esparcidos por el árbol, pero aún existen serios desacuerdos.
Por ejemplo, aún no está claro quiénes son los más cercanos en especie a los caracoles: moluscos bivalvos o moluscos
de patas de pala , dice Rokas. No se sabe exactamente cómo algunas de las primeras ramas de los animales de un árbol, como las medusas y las esponjas, están interconectadas. Los científicos pueden mostrar ejemplos de árboles en conflicto que aparecen en las mismas revistas científicas con una diferencia de semanas, o
incluso en el mismo número .
"De ahí la pregunta: ¿por qué es tan difícil para nosotros estar de acuerdo?" - dice Rokas.

Rokas y su estudiante graduado Leonidas Salichos estudiaron este tema
evaluando genes individualmente , utilizando los genes más útiles, que contienen la mayor cantidad de información relacionada con la historia evolutiva, para construir su versión del árbol.
Comenzaron con 23 especies de levadura y seleccionaron 1.070 genes. Para empezar, crearon un árbol filogenético de forma estándar, la concatenación. Para hacer esto, todas las secuencias de especies individuales se agrupan en un megagen, y luego las secuencias de especies individuales se comparan con esta secuencia larga, sobre la base de la cual se crea un árbol que explica mejor las diferencias.
El árbol resultante es preciso en términos de análisis estadístico estándar. Pero dado que métodos similares conducen a árboles repletos de desacuerdos, Rokas y Salichos decidieron profundizar en el tema. Construyeron conjuntos de árboles filogenéticos para genes de levadura individuales y aplicaron un algoritmo desarrollado utilizando la teoría de la información para buscar áreas de mayor correspondencia entre diferentes árboles. El resultado,
publicado en la revista Nature en mayo , fue inesperado. Cada gen estudiado parece contar una historia evolutiva ligeramente diferente.
"Casi todos los árboles construidos para genes individuales chocaron con un árbol basado en la concatenación de datos", dice Hilu. "Esto es impactante".
Llegaron a la conclusión de que si varios genes soportan una arquitectura particular, entonces debe ser precisa. Pero si diferentes conjuntos de genes soportan igualmente dos arquitecturas diferentes, entonces se reduce la probabilidad de que coincidan exactamente con la realidad. Rokas y Salichos usaron un método llamado
bootstrap estadístico para seleccionar los genes más informativos.
De hecho, "si solo toma genes con apoyo activo, obtendrá el árbol correcto", dice Donogue.
El árbol revisado coincidió con un árbol construido sobre una fuente alternativa de información evolutiva (cambios a gran escala en segmentos de ADN transmitidos de generación en generación) que justificaron su investigación.
Los descubrimientos no se limitaron a la levadura. Aplicando el mismo análisis a formas de vida más grandes y complejas, incluidos los datos genéticos de vertebrados y animales, encontraron serios conflictos entre genes individuales.
Algunos investigadores necesitan acostumbrarse a la idea de excluir selectivamente los datos del análisis. "Durante muchos años, el principal problema para las personas que intentan comprender las relaciones de los organismos ha sido el problema de recopilar suficientes datos", dice
Jeffrey Townsend , un biólogo evolutivo de Yale que no está involucrado en la investigación. "Siempre se ha informado a la comunidad sobre la necesidad de un conjunto de datos, por lo que no es sorprendente que hayan abordado la tarea de esa manera".
Aunque los biólogos evolucionistas han luchado con estos problemas durante años, el nuevo estudio se ha convertido en el mayor intento hasta la fecha para estudiar el nivel de conflicto de genes individuales. "La gente tendrá dos reacciones: hay más conflictos de lo que pensaba, y necesitamos aprender a analizarlos mejor", dice Donague, que quiere usar el nuevo método en su trabajo. Sin embargo, también señala dificultades para confirmar la precisión del nuevo enfoque. Aunque el árbol revisado coincide con lo que se basa en información genética alternativa, este último puede revelar sus propias inconsistencias. "No estoy seguro de saber cuál es realmente la relación", dice. "Y si no estamos seguros del verdadero estado de las cosas, no sabemos si obtuvimos el árbol correcto".
Cambio de imagen
Los investigadores necesitan aplicar la nueva técnica más ampliamente para ver cómo puede cambiar el concepto de evolución. Sin embargo, Rokas y Salichos ya han demostrado que es más difícil reconstruir ramas cortas del árbol, o partes "tupidas" del mismo, que representan períodos de especiación rápida, especialmente aquellos ubicados más cerca de la base del árbol y en lo profundo de la historia evolutiva.
"La investigación teórica predijo este comportamiento, pero nuestro estudio por primera vez demuestra la confirmación utilizando datos experimentales", dijo Rokas.
Rokas argumenta que los nuevos descubrimientos cambiarán la forma en que los investigadores interpretan partes de un árbol con marcos poco claros. “Los biólogos evolutivos generalmente asumen que si el árbol no tiene los detalles necesarios, entonces está mal. Y, por lo tanto, si recopilamos más datos y componimos mejores algoritmos, entonces llegaremos al árbol correcto ”, dice. Pero la presencia de partes conflictivas del árbol que persisten, a pesar de los flujos de datos y la aplicación de un nuevo tipo de análisis, puede indicar la presencia de partes espesas. "Creo que en algunos casos el algoritmo podrá resolver este conflicto, y en otros es posible marcar áreas de conflicto que es poco probable que podamos resolver".
El estudio de estas partes espesas del árbol puede dar una nueva mirada a las etapas especialmente interesantes de la evolución, por ejemplo, la explosión cámbrica, cuando la vida pasó del predominio de organismos simples a un conjunto abigarrado de especies animales.
Otros académicos coinciden en que los descubrimientos pueden influir en la forma en que los especialistas manejan las ideas en conflicto sobre la evolución. "Creo que esto es un presagio de un cambio de paradigma", dijo Townsend. "Si utilizamos métodos adecuados, tendremos la oportunidad de aprender más sobre los problemas que nos han afectado durante mucho tiempo".
Townsend, quien desarrolló su propio método para seleccionar los genes más informativos en función de
su ritmo evolutivo , señala que no todos los miembros de la comunidad científica están de acuerdo en la necesidad de nuevos enfoques. "Espero que este trabajo ayude a poner este tema en primer plano", dijo.
Elegir la cantidad correcta de genes para construir prototipos de árboles filogenéticos no es la única pregunta que afecta a los biólogos evolutivos. También deben acordar cuántas especies incluir en el procesamiento: cuantas más especies haya en el árbol, más difícil será el análisis. Los resultados también pueden variar debido a las diferencias en la calidad de los datos recopilados para diferentes especies. “Si necesitamos obtener una verdadera historia evolutiva de cómo todo está conectado entre sí, entonces, ¿qué es mejor para esto: recolectar más genes o más especies? - dice Donogue. "Creo que ambos".
Los nuevos enfoques que permiten a los investigadores obtener resultados precisos utilizando menos genes pueden expandir el árbol evolutivo. La capacidad de seleccionar solo los genes más informativos puede hacer que el proceso sea más eficiente y permitir a los científicos crear árboles precisos utilizando menos datos y recursos. "Si pudiéramos seleccionar varios genes y obtener el mismo árbol bueno que con todo el genoma", dice Khilu, "podríamos construir un árbol de la vida mucho más detallado - a nivel de géneros, o incluso a nivel de especies - en lugar de contentarnos con el esqueleto de las ramas más importantes ".