
A pesar de la existencia de obvios obstáculos y dificultades que a veces se interponen en el desarrollo y la implementación de productos de ingeniería genética (IG), el siglo XXI no se puede imaginar sin los frutos de esta importante y diversa tecnología en el arsenal de un biólogo moderno. El organismo más utilizado en el GI es la bacteria.
¿Qué es GI y por qué lo necesitamos? ¿Por qué las bacterias son tan populares entre los ingenieros genéticos? ¿De qué forma es la forma más fácil de introducir el gen deseado en la bacteria? ¿Qué dificultades se pueden encontrar al trabajar con estos organismos? ¿Qué sucedió antes: la creación de la primera bacteria genéticamente modificada o el descubrimiento de la estructura del ADN y el genoma? Lea sobre esto y mucho más debajo del gato.
0. Breve programa educativo en biología
Este párrafo proporciona una breve descripción del llamado
Dogma central de la biología molecular . Si tiene conocimientos básicos de biología molecular, no dude en pasar al paso 1.
 El dogma central de la biología molecular en una imagen.
El dogma central de la biología molecular en una imagen.Entonces comencemos. Toda la información sobre todas las etapas de desarrollo y las propiedades de cualquier organismo, ya sea
procariotas (bacterias),
arqueas o
eucariotas (el resto son simples y multicelulares), está codificada en el ADN genómico, que es un complejo de dos cadenas polinucleotídicas complementarias entre sí, formando una doble hélice ( nucleótidos de ADN complementarios: AT y GC). Los cromosomas eucariotas son moléculas de ADN lineales de doble cadena, y los cromosomas procariotas están en bucle. A menudo, los genes constituyen solo una pequeña parte de todo el genoma (en humanos, alrededor del 1.5%).
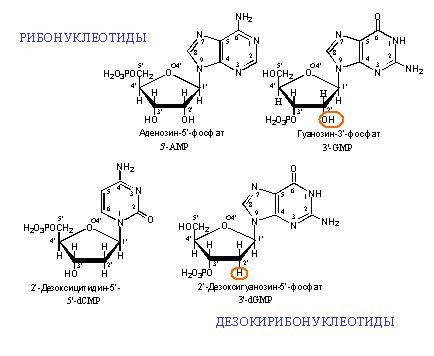 Ejemplos de monómeros de ADN y ARN. "Deoxy" en el nombre del ADN significa la ausencia de un átomo de oxígeno en la posición 2 '(en la figura, la posición 2' está encerrada en rojo).
Ejemplos de monómeros de ADN y ARN. "Deoxy" en el nombre del ADN significa la ausencia de un átomo de oxígeno en la posición 2 '(en la figura, la posición 2' está encerrada en rojo).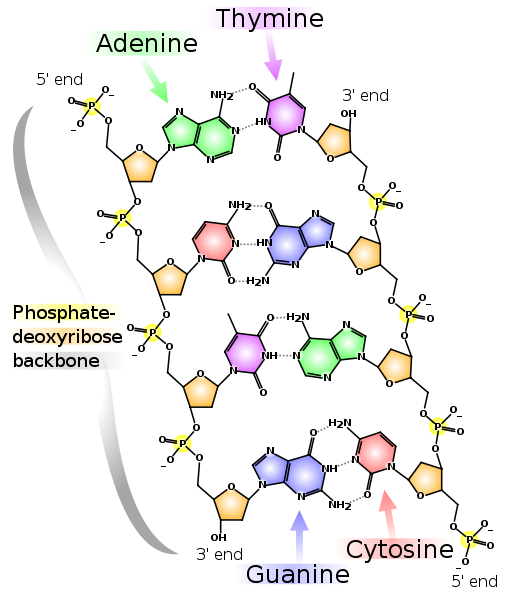 Dos cadenas de ADN complementarias. Las líneas discontinuas muestran los enlaces de hidrógeno entre las bases. Como se puede ver, la adenina y la timina forman dos enlaces de hidrógeno entre sí, y la guanina y la citosina forman tres. Por lo tanto, el enlace GC es más fuerte y las secciones ricas en GC de ADN bicatenario son más difíciles de separar en dos cadenas.
Dos cadenas de ADN complementarias. Las líneas discontinuas muestran los enlaces de hidrógeno entre las bases. Como se puede ver, la adenina y la timina forman dos enlaces de hidrógeno entre sí, y la guanina y la citosina forman tres. Por lo tanto, el enlace GC es más fuerte y las secciones ricas en GC de ADN bicatenario son más difíciles de separar en dos cadenas.Tenga en cuenta que cada una de las cadenas tiene un extremo 5 'y un extremo 3'. Se puede ver que cerca del extremo 5 'de la cadena izquierda está el extremo 3' de la derecha y viceversa, por lo tanto, las cadenas se llaman "antiparalelas". El ARN también tiene un extremo 5 'y 3'. Las posiciones 5 'y 3' se eligieron para indicar el principio y el final porque es a través de ellas que se forman enlaces covalentes en las cadenas de ADN y ARN.
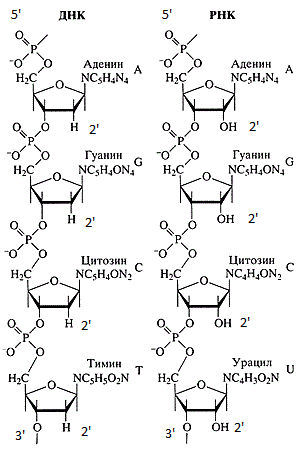 Cadenas de ADN y ARN.
Cadenas de ADN y ARN.Las secuencias de ADN y ARN siempre se registran desde el extremo 5 'hasta el extremo 3'. Hay varias razones para esto:
- La síntesis de nuevas cadenas de ADN y ARN comienza desde el extremo 5 '( ADN polimerasa (enzimas que sintetizan una cadena de ADN complementaria en una matriz de ADN o ARN) y ARN polimerasa (enzimas que sintetizan una cadena de ARN complementaria en una matriz de ADN o ARN) dirección 3 '-> 5', por lo que se sintetiza una nueva cadena en la dirección 5 '-> 3');
- El ribosoma lee los codones, moviéndose a lo largo del ARNm en la dirección de 5 '-> 3';
- La secuencia de aminoácidos se escribe en la cadena de codificación de ADN en la dirección 5 '-> 3' (una parte significativa del ARNm es una copia exacta de la región de codificación de ADN con timina reemplazada por uracilo y con un grupo hidroxilo (-OH) en lugar de hidrógeno en la posición 2 ', por supuesto);
- Finalmente, es conveniente tener una regla de grabación generalmente aceptada.
Un gen es una porción de ADN genómico que define la secuencia de nucleótidos de una molécula de ARN:
- ARN codificador: ARN mensajero (ARNm), en el que la secuencia de aminoácidos de la proteína correspondiente se codifica como codones. También puede encontrar el nombre "ARN informativo", luego la abreviatura se parece a "ARNm";
- ARN no codificante: ARN de transporte, ARN ribosómico y otros.
El papel del ARNt es entregar aminoácidos al complejo de ARNm-ribosoma. Además, es el ARNt el responsable del reconocimiento de los codones de ARNm; para esto, cada ARNt incluye el llamado "anticodón", un triplete complementario al codón de ARNm.
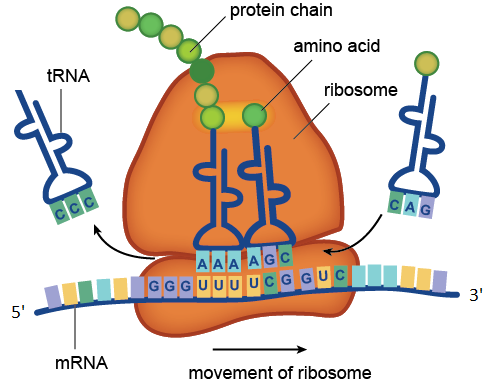 El proceso de traducción catalizado por el ribosoma. En la figura, los codones UUU y UCG contenidos en el ARNm son reconocidos por los anticodones AAA y AGC contenidos en las moléculas de ARNt. El ARN de transporte con anticodón CCC ya ha dado su aminoácido a la cadena de proteínas en crecimiento, y el ARNt con anticodón CAG está esperando en la cola. La gráfica de la molécula de ARNm que se muestra en la figura consta de cuatro codones: GGGUUUUCGGUC. El codón GGG corresponde al aminoácido glicina, UUU a fenilalanina, UCG a serina, GUC a valina. Entonces, esta región de ARNm codifica un fragmento de proteína con la secuencia de aminoácidos glicina-fenilalanina-serina-valina.
El proceso de traducción catalizado por el ribosoma. En la figura, los codones UUU y UCG contenidos en el ARNm son reconocidos por los anticodones AAA y AGC contenidos en las moléculas de ARNt. El ARN de transporte con anticodón CCC ya ha dado su aminoácido a la cadena de proteínas en crecimiento, y el ARNt con anticodón CAG está esperando en la cola. La gráfica de la molécula de ARNm que se muestra en la figura consta de cuatro codones: GGGUUUUCGGUC. El codón GGG corresponde al aminoácido glicina, UUU a fenilalanina, UCG a serina, GUC a valina. Entonces, esta región de ARNm codifica un fragmento de proteína con la secuencia de aminoácidos glicina-fenilalanina-serina-valina.Los ARN ribosómicos son componentes indispensables del ribosoma. La función principal del ARNr es garantizar el proceso de traducción: está involucrado en la lectura de información del ARNm utilizando moléculas adaptadoras de ARNt y catalizando la formación de enlaces peptídicos entre los aminoácidos unidos al ARNt y la cadena de proteínas en crecimiento.
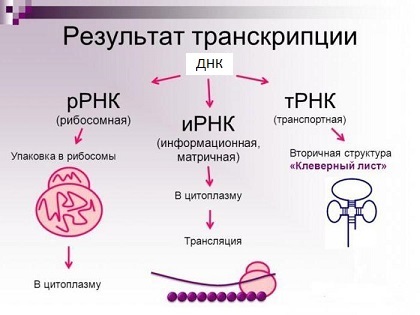 Los principales tipos de moléculas de ARN (de hecho, hay mucho más).
Los principales tipos de moléculas de ARN (de hecho, hay mucho más).Una proteína, por otro lado, es una cadena de aminoácidos unidos covalentemente a través de un enlace peptídico (puede ver un poco más de lo que está en el spoiler). Después de la síntesis, la cadena de aminoácidos debería adoptar una determinada estructura espacial: "
conformación " (
ya me contaron sobre la estructura espacial de las proteínas
en Geektimes ). Además, muchas proteínas grandes en realidad consisten en varias proteínas combinadas por interacciones hidrófobas y enlaces de hidrógeno en una sola estructura estable. En este caso, cada una de las "proteínas de construcción" se denomina "subunidad", y la proteína grande resultante se denomina "multisubunidad".
20 aminoácidos que forman proteínas  Complejo ribosómico. La imagen fue tomada de la publicación OlegKovalevskiy "Impresión 3D de modelos de moléculas de proteínas" .
Complejo ribosómico. La imagen fue tomada de la publicación OlegKovalevskiy "Impresión 3D de modelos de moléculas de proteínas" .En el caso de los genes que codifican una proteína, el proceso de decodificación de la información genética se ve así:
- La ARN polimerasa reconoce al promotor y se une a él (si está "abierto", discutiremos más a fondo la regulación de la actividad del promotor);
- En la matriz de ADN, la enzima ARN polimerasa, de acuerdo con el principio de complementariedad, sintetiza el "blanco" de la matriz ARN (pre-ARNm, en eucariotas) o el ARNm funcional terminado (en procariotas). Este proceso se llama "transcripción" ;
- (solo en eucariotas) La molécula pre-ARNm sufre modificaciones ("madura") y se convierte en ARNm funcional;
- El ARNm es reconocido por el ribosoma , una enzima que decodifica el código de tripletes del ARNm y, en base a él, sintetiza un péptido / proteína. Los aminoácidos a partir de los cuales el ribosoma construye la proteína se entregan en complejo con ARN de transporte ( ARNt ). Este proceso se llama "difusión" ;
- El péptido / proteína puede sufrir modificaciones postraduccionales ("maduración" por analogía con ARNm) y se vuelve funcional. Un factor importante es que el sistema de modificación postraduccional de eucariotas es mucho más complejo y diverso que el de los procariotas, por lo tanto, no todas las proteínas eucariotas pueden ser sintetizadas correctamente por una bacteria.
Además de las regiones de codificación, el genoma contiene numerosos fragmentos que también participan en la transcripción de una forma u otra. Las parcelas ubicadas cerca del gen y llamadas promotores son reconocidas por las ARN polimerasas (dicen que el gen está bajo el control de este promotor). Diferentes promotores son reconocidos por diferentes ARN polimerasas. Por ejemplo, un gen bajo el control de un promotor
bacteriófago no se transcribirá en bacterias si la ARN polimerasa del bacteriófago correspondiente no se sintetiza en él.
En generalCada gen también puede tener varias secuencias reguladoras, que pueden ubicarse directamente cerca del promotor (o incluso superponerse con él) o a una distancia de decenas de miles de pares de nucleótidos. Los elementos potenciadores de la transcripción se denominan
"potenciadores", los elementos supresores de la transcripción se denominan silenciadores, y las proteínas que interactúan con ellos se denominan
factores de transcripción . Aunque también es habitual llamar a los factores de transcripción los componentes necesarios del complejo de iniciación de la transcripción, sin los cuales la transcripción es imposible en principio. El hecho es que solo para comenzar la síntesis de la molécula de ARN en la matriz de ADN en eucariotas y arqueas, es necesario el ensamblaje de todo el complejo supramolecular. El complejo más simple incluye la holoenzima ARN polimerasa y seis denominados
"factores de transcripción comunes" (TFIIA, TFIIB, TFIID, TFIIE, TFIIF y TFIIH). El complejo en sí se llama el
"complejo de preiniciación de transcripción" (
video , cada componente del complejo se resalta en un color u otro).
El complejo de transcripción procariota es completamente diferente, por lo que no tiene sentido incrustar el gen eucariota con el promotor eucariota en la bacteria. Un análogo procariota de los factores de transcripción comunes de eucariotas y arqueas se puede llamar una proteína llamada
"factor sigma" .
 Complejo transcripcional procariota. Las letras que se muestran en la figura son designaciones generalmente aceptadas de las subunidades correspondientes. σ70 - factor sigma de genes domésticos de E. coli
Complejo transcripcional procariota. Las letras que se muestran en la figura son designaciones generalmente aceptadas de las subunidades correspondientes. σ70 - factor sigma de genes domésticos de E. coliLos genomas de procariotas y eucariotas tienen muchas características en común, y el dogma central de la biología molecular, mencionado anteriormente, es cierto para ambos reinos. Sin embargo, también hay muchas diferencias significativas. Por ejemplo, una bacteria se caracteriza por un sistema de operones: genes agrupados que participan en el mismo proceso y no se transcriben por separado, sino como parte de un ARNm largo. En eucariotas, todo es completamente diferente: los genes involucrados en un proceso están dispersos en diferentes cromosomas, y los genes mismos se dividen en fragmentos
de codificación
de exones por regiones
de intrones no codificantes. En este caso, al principio el gen se transcribe completamente, y luego, ya en la etapa de ARN, los intrones se extirpan y los exones se entrecruzan para formar el ARNm de codificación. Este proceso se llama
empalme . Al mismo tiempo, no todos los exones disponibles se pueden unir en el ARNm terminado, pero solo una parte de ellos, en este caso, habla de
"empalme alternativo" . Por lo tanto, una célula eucariota puede sintetizar varias proteínas, mientras transcribe el mismo gen. Entre otras cosas, esto da una consecuencia muy importante: a menudo no tiene sentido insertar el gen eucariota en la bacteria "como está en el cromosoma", ya que la bacteria simplemente no puede empalmarse.
Hay otra diferencia importante. Los procariotas se caracterizan por la presencia de material genético basado en el ADN fuera del anillo "cromosoma", los llamados
"plásmidos" , pequeñas moléculas circulares de ADN de doble cadena. Además, los procariotas carecen de orgánulos, incluido el núcleo: todos los componentes de una célula bacteriana pueden viajar libremente a través del espacio intracelular. Los eucariotas, sin embargo, no tienen plásmidos, pero hay
plastidios y
mitocondrias en el genoma de los cuales se incluyen los plásmidos (de acuerdo con la hipótesis más justificada, los plastidios y las mitocondrias son "descendientes" de la arquitectura procariótica del genoma de las cianobacterias y bacterias atrapadas en el interior por antiguos protoeukaryotes unicelulares). Además, la presencia de un núcleo y otros compartimentos intracelulares rodeados por su propia membrana ya es típica de los eucariotas. Por lo tanto, la ingeniería genética de las células eucariotas requiere enfoques diferentes que la ingeniería genética de las bacterias.
El código genético en sí está estructurado de la siguiente manera. Cada gen / exón consiste en un conjunto de tripletes / codones, secuencias de tres nucleótidos entre los cuales no hay espacios. La organización triplete es válida tanto para los genes en el ADN como para la parte de codificación del ARNm. En el proceso de traducción, los ARN de transporte (ARNt) que transportan un aminoácido específico "reconocen" sus trillizos de tres letras correspondientes. El ribosoma desconecta el aminoácido del ARNt y lo une a la cadena de aminoácidos en crecimiento, que al final de la traducción se convertirá inmediatamente en una proteína madura, completamente funcional, o antes de que sufra una serie de modificaciones adicionales. En este caso, solo un aminoácido corresponde a cada triplete, pero varios codones diferentes pueden corresponder a un aminoácido. Esto es comprensible, porque en el código genético estándar hay 61 codones codificadores, y
solo hay
20 aminoácidos proteinogénicos (codones totales, por supuesto, 4 * 4 * 4 = 64, pero tres de ellos no codifican, en cambio, sirven como una señal para detener la traducción y se llaman " detener los codones ").
 Codones en el código genético estándar. Gracias a Wikipedia por la foto.
Codones en el código genético estándar. Gracias a Wikipedia por la foto.Entonces, las proteínas son precisamente esos mismos elementos que son el último eslabón en la cadena entre el ADN genómico y las propiedades del cuerpo, el llamado
"fenotipo" . Por lo tanto, para cambiar de alguna manera las características del organismo que es importante para nosotros, necesitamos cambiar su ADN de tal manera que, como resultado, ciertas proteínas aparezcan en sus células, lo que nos proporcionará el resultado objetivo. Esta es la idea básica de toda la ingeniería genética.
1) ¿Para qué se usan las bacterias en la ingeniería genética y por qué
Entonces, descubrimos cómo y por qué la secuencia de ADN genómico afecta las propiedades y características del cuerpo. Por supuesto, será muy bueno si el rasgo está completamente determinado por un solo gen; insertar un pequeño fragmento ya no es un problema grave. Por ejemplo, a menudo la resistencia de una planta a un herbicida o plaga está determinada por un solo gen, por lo que crear variedades con la resistencia deseada en tales casos no es difícil (en oposición a llevar dicha planta al mercado). Lo mismo es cierto para muchas resistencias a los antibióticos de las bacterias (de hecho, las bacterias tienen muchos mecanismos de protección contra los antibióticos, pero funcionan de manera independiente). El ejemplo opuesto es, por ejemplo, un intento de los científicos de enseñar a las plantas a absorber el nitrógeno de la atmósfera. El hecho es que la única fuente de nitrógeno para las plantas es el suelo en el que los compuestos que contienen nitrógeno adecuados para la asimilación por la planta son sintetizados por microorganismos (o introducidos en forma de fertilizantes por un jardinero o un perro que pasa). Obviamente, crear una planta con un mecanismo nutricional alternativo sería muy beneficioso para la agricultura. Pero, desafortunadamente, este proceso es tan complicado que el problema de su "transferencia" del microorganismo a la planta no se ha resuelto hasta ahora.
Finalmente, si nuestro objetivo es obtener proteínas para cualquier propósito específico (estudiar la estructura y funciones de la proteína, crear preparaciones médicas o reactivos de laboratorio basados en ella, etc.), entonces, obviamente, también estamos muy contentos con la integración de un solo gen en la célula, que en este caso, se acostumbra llamar al "organismo productor".
Una bacteria en la ingeniería genética es un material fuente potencial para crear:
- un productor de la proteína que necesitamos a escala de laboratorio o industrial;
- un agente activo en una cierta transformación química de un compuesto en otro, ya sea un proceso de fermentación en la industria alimentaria, la creación de condiciones más favorables para el crecimiento de las plantas mediante la introducción de un "productor de fertilizantes bacterianos" en el suelo o la utilización de chatarra de acero;
- klonotek de genes (un tema, cuya buena descripción aumentará el tamaño del artículo a indecente);
- un medicamento médicamente significativo, por ejemplo, para restaurar la microflora del tracto gastrointestinal;
- cepas bacterianas de Agrobacterium tumefaciens para la posterior modificación genética de las plantas.
* Podría olvidar algo, por lo que las adiciones en los comentarios son bienvenidas.
Un hecho interesante es que los primeros experimentos exitosos en el campo de la ingeniería genética de bacterias ocurrieron mucho antes del trabajo histórico de Watson y Crick. Además, sobre la base de estos experimentos, se demostró el hecho mismo de que la información está contenida en el ADN, después de lo cual los científicos no pudieron dedicar su tiempo a las hipótesis sobre el ARN y las proteínas.
Este trabajo, realizado en 1944, se conoce como el
Experimento Avery, MacLeod y McCarthy , basado en el
trabajo de Frederick Griffith , durante el cual se descubrió que la infección de cepas de neumococos patógenos muertos y no patógenos vivos causa el desarrollo de la enfermedad, mientras que individualmente no causan síntomas significativos. De este experimento, se concluyó que una bacteria muerta es capaz de transmitir algo a un "colega" no patógeno, como resultado de lo cual se vuelve peligroso. ¿Pero qué se pasan el uno al otro? Para 1944, había tres candidatos principales: ADN, ARN y proteínas. Para establecer el portador, se llevó a cabo un elegante experimento: en ese momento, las enzimas capaces de destruir por separado el ADN (DNasa), el ARN por separado (RNasa) y las proteínas por separado (proteinasa) ya estaban disponibles. Se demostró que la transferencia de propiedades patogénicas no se produjo solo en aquellos casos en que la preparación de una cepa patógena muerta se trató con DNasa y no dependía del tratamiento del fármaco con RNasa y proteinasa.
Por lo tanto, se demostró que el ADN es el portador de información sobre los signos. Además, se demostró claramente que es posible la penetración espontánea de una molécula de ADN extraña en una célula bacteriana.
¿Por qué las bacterias son tan populares con defectos obvios (por ejemplo, la falta de modificaciones eucarióticas postraduccionales)? Todo es simple
Operan sin pretensiones, son fáciles de usar y no requieren medios nutritivos costosos.2) ¿Cómo se crea una construcción genética que se introduce en la bacteria?
La ingeniería genética moderna de las bacterias consiste principalmente en la introducción de un vector plasmídico (un plásmido bacteriano modificado que contiene el gen objetivo y un conjunto de otros elementos necesarios, que se analizarán a continuación). Cambiar el cromosoma bacteriano es menos típico, pero este procedimiento tampoco es descabellado: por ejemplo, el gen de la ARN polimerasa del bacteriófago T7 se introdujo en el cromosoma de E. coli utilizando un vector basado en el profágico λ durante la creación de una de las cepas populares en la placa de laboratorio. Hay tres razones por las cuales un investigador a menudo elige introducir un gen en un vector plasmídico:Un vector plasmídico típico para trabajar con bacterias es una molécula de ADN de doble cadena circular que lleva el gen de la proteína objetivo bajo el control de un promotor específico y una cantidad de genes y elementos reguladores necesarios, cuya presencia asegura una cantidad constante de plásmido en la célula ("control de copia"). Obviamente, incluso en el caso de síntesis ultraeficiente de ARNm, el vector es de poca utilidad si existe en la bacteria en la cantidad de un par de piezas: en el proceso de división, la probabilidad de formación de células hijas sin el plásmido necesario será banal.Además del gen y el promotor, los elementos principales del vector plasmídico son:- ori es la región de origen de la replicación del plásmido. Necesario para mantener una cantidad constante de plásmido y su herencia por las células hijas;
- — , , , . , , (« »). , , . , .
, β- (GUS). , . , . — (GFP) ( GUS GFP );
- , ( — , — );
- — , ( ). , «» .
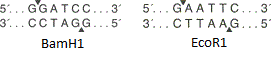 La figura muestra los sitios de endonucleasa de restricción BamH1 y la EcoR1 . Ambas enzimas reconocen un sitio específico de seis pares de bases e introducen roturas monocatenarias en diferentes lugares (indicados por flechas triangulares). En este caso, los puntos de ruptura de la cadena no coinciden, lo que significa que se forman "extremos adhesivos" (si coinciden, se forman "extremos romos").
La figura muestra los sitios de endonucleasa de restricción BamH1 y la EcoR1 . Ambas enzimas reconocen un sitio específico de seis pares de bases e introducen roturas monocatenarias en diferentes lugares (indicados por flechas triangulares). En este caso, los puntos de ruptura de la cadena no coinciden, lo que significa que se forman "extremos adhesivos" (si coinciden, se forman "extremos romos").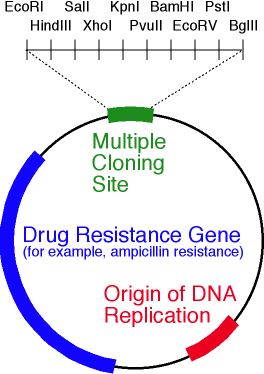 Un esquema simplificado de un vector plasmídico. La figura muestra ori, el gen de resistencia a los antibióticos y el polienlazador que contiene 10 sitios de endonucleasa de restricción.Bueno, el vector está en nuestras manos. ¿Cómo incrustar un gen en él? Y de todos modos, ¿dónde puedo obtener este gen?Supongamos que conocemos la secuencia de nucleótidos del gen que necesitamos. Luego proceda de la siguiente manera:
Un esquema simplificado de un vector plasmídico. La figura muestra ori, el gen de resistencia a los antibióticos y el polienlazador que contiene 10 sitios de endonucleasa de restricción.Bueno, el vector está en nuestras manos. ¿Cómo incrustar un gen en él? Y de todos modos, ¿dónde puedo obtener este gen?Supongamos que conocemos la secuencia de nucleótidos del gen que necesitamos. Luego proceda de la siguiente manera:- , ;
- .
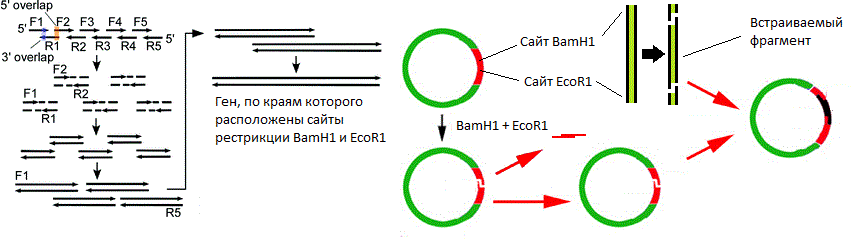 ( ). .Los cebadores superpuestos se sintetizan de tal manera que después del ensamblaje, se forma un gen de tamaño completo, en cuyos lados hay sitios de corte con las mismas endonucleasas, cuyos sitios se encuentran en el polienlazador del vector. Por lo tanto, si las endonucleasas necesarias de proceso (en mi ejemplo son enzimas BamH1 y la EcoR1 ) vector y el gen se ensambla, a continuación, se forman extremos pegajosos que pueden "conocen entre sí" debido a las interacciones de nucleótidos complementarios extremos pegajosos. Pero el reconocimiento por sí solo no es suficiente, porque hasta ahora los extremos adhesivos se mantienen entre sí únicamente debido a los frágiles enlaces de hidrógeno. Este problema se resuelve agregando a la mezcla de reacción simultáneamente el vector cortado, el gen cortado y la enzima ADN ligasa ., que elimina las roturas de la cadena en las moléculas de ADN de doble cadena.Otro factor importante en el ensamblaje de genes es el hecho de que las frecuencias de uno u otro codón son diferentes para diferentes organismos, mientras que en las células generalmente hay más de esos tRNA que corresponden a codones más "populares". Dado que muchos aminoácidos están codificados por varios codones, es muy probable que al copiar un gen de un organismo a otro sin pensarlo, corramos el riesgo de sufrir un gran retraso en el proceso de traducción. De hecho, aunque muchos codones de este gen son raros en el nuevo organismo, el ribosoma esperará más tiempo cuando finalmente llegue el ARNt deseado.
( ). .Los cebadores superpuestos se sintetizan de tal manera que después del ensamblaje, se forma un gen de tamaño completo, en cuyos lados hay sitios de corte con las mismas endonucleasas, cuyos sitios se encuentran en el polienlazador del vector. Por lo tanto, si las endonucleasas necesarias de proceso (en mi ejemplo son enzimas BamH1 y la EcoR1 ) vector y el gen se ensambla, a continuación, se forman extremos pegajosos que pueden "conocen entre sí" debido a las interacciones de nucleótidos complementarios extremos pegajosos. Pero el reconocimiento por sí solo no es suficiente, porque hasta ahora los extremos adhesivos se mantienen entre sí únicamente debido a los frágiles enlaces de hidrógeno. Este problema se resuelve agregando a la mezcla de reacción simultáneamente el vector cortado, el gen cortado y la enzima ADN ligasa ., que elimina las roturas de la cadena en las moléculas de ADN de doble cadena.Otro factor importante en el ensamblaje de genes es el hecho de que las frecuencias de uno u otro codón son diferentes para diferentes organismos, mientras que en las células generalmente hay más de esos tRNA que corresponden a codones más "populares". Dado que muchos aminoácidos están codificados por varios codones, es muy probable que al copiar un gen de un organismo a otro sin pensarlo, corramos el riesgo de sufrir un gran retraso en el proceso de traducción. De hecho, aunque muchos codones de este gen son raros en el nuevo organismo, el ribosoma esperará más tiempo cuando finalmente llegue el ARNt deseado. , , , , , . E. coli .Ahora unas palabras sobre el promotor. La selección de un promotor adecuado es muy importante, ya que el proceso de transcripción depende en gran medida de ello. Los promotores se dividen condicionalmente en fuertes, medios y débiles. La "fuerza" del promotor está determinada por cuán activamente se transcriben los genes bajo su control, todas las demás cosas son iguales: cuanto más activa sea la transcripción, más fuerte será el promotor. Obviamente, cuando queremos crear un productor de proteínas, debemos comenzar con fuertes promotores. En algunos casos, la transcripción excesivamente rápida (por lo tanto, la traducción activa) daña la célula, en cuyo caso puede intentar usar un promotor más débil. Aunque, de hecho, es mucho más fácil influir en la actividad de transcripción de un productor existente que crear uno nuevo.Otra cosa es importante. A menudo, las proteínas codificadas por el vector tienen un efecto extremadamente negativo sobre la viabilidad bacteriana. La síntesis de estas proteínas no solo ocupa una gran cantidad de recursos (y la cantidad de proteína objetivo debe ser buena, no menos del 10% del peso seco total de la célula), sino que también flotan de un lado a otro en el citoplasma con una carga muerta . Por lo tanto, por el momento, es mejor desactivar por completo la expresión de un gen extraño a la célula. Para este propósito, se han desarrollado sistemas de expresión controlada que le permiten "habilitar" la expresión del gen que necesitamos "a pedido". Los más comunes son:
, , , , , . E. coli .Ahora unas palabras sobre el promotor. La selección de un promotor adecuado es muy importante, ya que el proceso de transcripción depende en gran medida de ello. Los promotores se dividen condicionalmente en fuertes, medios y débiles. La "fuerza" del promotor está determinada por cuán activamente se transcriben los genes bajo su control, todas las demás cosas son iguales: cuanto más activa sea la transcripción, más fuerte será el promotor. Obviamente, cuando queremos crear un productor de proteínas, debemos comenzar con fuertes promotores. En algunos casos, la transcripción excesivamente rápida (por lo tanto, la traducción activa) daña la célula, en cuyo caso puede intentar usar un promotor más débil. Aunque, de hecho, es mucho más fácil influir en la actividad de transcripción de un productor existente que crear uno nuevo.Otra cosa es importante. A menudo, las proteínas codificadas por el vector tienen un efecto extremadamente negativo sobre la viabilidad bacteriana. La síntesis de estas proteínas no solo ocupa una gran cantidad de recursos (y la cantidad de proteína objetivo debe ser buena, no menos del 10% del peso seco total de la célula), sino que también flotan de un lado a otro en el citoplasma con una carga muerta . Por lo tanto, por el momento, es mejor desactivar por completo la expresión de un gen extraño a la célula. Para este propósito, se han desarrollado sistemas de expresión controlada que le permiten "habilitar" la expresión del gen que necesitamos "a pedido". Los más comunes son:- Un sistema basado en elementos reguladores del operón lactosa E. coli ( lac -peron) y un fuerte promotor.
El hecho es que E. coli tiene sus propias reglas nutricionales. En primer lugar, existe un mecanismo para suprimir la actividad del lac -peron, que se activa solo cuando la lactosa no ingresa a la célula. Esto es lógico: ¿por qué desperdiciar energía en la síntesis de lo que no es útil? Pero tan pronto como la lactosa comienza a ingresar a la célula en cantidades suficientes, este mecanismo se apaga.
Sin embargo, hay un segundo mecanismo para suprimir la actividad del lac -peron. Si hay glucosa en el medio, entonces la célula se alimenta exclusivamente de glucosa, ya que activa el segundo mecanismo de inhibición de la transcripción del lac -peron. Por lo tanto, el lac -peron está activo solo cuando solo hay lactosa en el espacio que rodea la célula. El menos del operón de lactosa es un promotor extremadamente débil, por lo tanto, en las cepas productoras se reemplaza por uno fuerte. Los promotores fuertes a menudo se derivan de los patógenos. Los promotores más fuertes más utilizados en la ingeniería genética de los procariotas están aislados de virus bacterianos: bacteriófagos . Por ejemplo, el promotor del fago T7 se usa ampliamente.
Por cierto, algunos promotores fuertes para la ingeniería genética de las plantas también están aislados de los virus, por ejemplo, este es el promotor del virus del mosaico de la coliflor.

Como se mencionó anteriormente, E. coli no tiene una ARN polimerasa que reconozca los promotores de bacteriófagos; por lo tanto, el gen de ARN polimerasa del bacteriófago correspondiente se inserta previamente en el productor.
El popular sistema de síntesis de proteínas a base de E. coli lleva el gen de la ARN polimerasa del fago T7 bajo el control del promotor bacteriano de la ARN polimerasa, regulado por el mecanismo de la lactona . Si esta cepa se transforma con un vector que lleva el gen objetivo bajo el control del "promotor fago T7 + regulación del complejo promotor tipo lacperón ", entonces surgirá un mecanismo de dos niveles para inhibir la transcripción del gen objetivo.
Cuando se usa este diseño, la glucosa y la lactosa se agregan al medio nutriente simultáneamente. Durante algún tiempo, las células se alimentarán de glucosa y se dividirán en silencio, ya que la síntesis de una proteína extraña se suprime por completo. Cuando termine la glucosa y las células cambien al metabolismo de la lactosa, ya habrá suficiente biomasa en el cultivo, es el momento de comenzar la síntesis de la proteína que necesitamos. Este procedimiento se llama "autoinducción".
Puede hacerlo de otra manera: no agregue glucosa y lactosa al medio nutriente, y luego, cuando el cultivo alcance la densidad deseada, agregue lo que la célula tomará para la lactosa, pero no puede metabolizarlo ni destruirlo. Ahora, IPTG se usa como tal inductor.
- Un sistema basado en el mecanismo regulador del promotor pL del bacteriófago λ .
Este promotor es inactivado por la proteína represora cI. En este caso, se descubrió una forma termosensible de esta proteína llamada cI857: este factor de transcripción conserva la funcionalidad a una temperatura de aproximadamente 30 ° C y la pierde a 42 ° C. Por lo tanto, cuando se usa dicho sistema, el cultivo bacteriano se cultiva primero a la densidad deseada a 30 ° C, y luego la temperatura se eleva a 42 ° C, comenzando así la síntesis de la proteína objetivo.
Bueno, el vector está diseñado. Entonces lo pequeño es encontrar un método adecuado para su introducción en la célula bacteriana. Pero esta es una historia completamente diferente.