El análisis de la afiliación de la población humana por ADN, en nuestra experiencia, plantea tres grandes preguntas entre el público: ¿pueden vincularse los genes y los grupos étnicos, cómo es el análisis del origen desde un punto de vista técnico y si las pruebas genéticas pueden "identificar a los judíos"? Por alguna razón, es precisamente el tema de la identidad judía con el ADN lo que preocupa tanto a quienes tienen evidencias innegables de pertenecer a las personas elegidas por Dios como a quienes no comen matzá y no leen la Torá.
En el nuevo material de Genotek en Geektimes, trataremos de responder todo en orden. Y sí, también definimos judíos.

Razas también conocidas como grupos de población en biología, medicina y genética.
La humanidad tiene el mal hábito de justificar la violencia con la superioridad "innata" de una raza sobre otra; es por eso que los biólogos modernos abordan el tema de las diferencias genéticas entre las poblaciones con precaución. La (no) existencia de límites biológicos entre los grupos raciales y étnicos se ha discutido vehementemente durante todo el siglo XX, pero aún no se ha alcanzado un consenso final sobre este tema (
1 ).
Se esperaba que la secuenciación del genoma humano uniera a todos. El genoma, leído "de" y "a", mostrará que los límites entre los grupos son de naturaleza social y que los genes son los mismos para todos. Resultó diferente: un estudio cuidadoso del código de nucleótidos humanos revivió y aumentó el interés en las diferencias biológicas entre las poblaciones raciales y étnicas. Los mismos genes, en general, encontraron variantes alélicas ligeramente diferentes asociadas con el riesgo de enfermedades (
2 ), el metabolismo de los medicamentos (
3 ), la respuesta del cuerpo a las condiciones ambientales (
4 ), y estas variantes se encontraron en diferentes poblaciones con diferentes frecuencias.
La búsqueda de genes "indios" o "africanos" inexistentes se ha detenido, pero la investigación en el campo de la genética médica y de la población aún establece paralelismos entre las características biológicas y el origen étnico de los participantes. El uso de los términos "raza" y "etnia" en tales trabajos se discute activamente (y a menudo se condena). Hubo intentos de introducir reglas que obligaran a los investigadores a justificar la necesidad de utilizar categorías "resbaladizas" y aclarar qué se entiende exactamente por términos específicos. En febrero del año pasado, Science, una de las revistas científicas más respetadas, publicó un artículo ambiguo (
5 ), proponiendo abandonar por completo el uso del término "raza" en la investigación genética, reemplazándolo por una "ascendencia" más correcta y neutral: .
Pero incluso en condiciones de incertidumbre con los términos, la humanidad todavía está dividida en grupos de población: en particular, para la correcta realización de ensayos clínicos de drogas y la evaluación del riesgo de enfermedades. Por ejemplo, tres variantes alélicas del gen NOD2 - R702W, G908R y 1007fs - están asociadas con un mayor riesgo de enfermedad de Crohn en los europeos europeos (
6 ,
7 ), sin embargo, ninguna de estas variantes está asociada con la enfermedad de Crohn en japonés (
8 ). Se sabe que los alelos del gen CCR5 afectan la tasa de desarrollo de inmunodeficiencia en pacientes infectados con VIH (
9 ): entre ellos, se encontró una opción que ralentiza la progresión de la enfermedad en los estadounidenses de ascendencia europea, pero acelera su desarrollo en los afroamericanos (
10 ). Los asiáticos encontraron una correlación entre los polimorfismos del gen de la proteína p53, que regula la respuesta al estrés y suprime el desarrollo de tumores, y las temperaturas promedio de invierno en los hábitats de las poblaciones: adaptación genética a las heladas (
11 ). Y si en el pasado solo se utilizaba la información proporcionada por los propios participantes para dividir la muestra en grupos étnicos, en la era postgenómica se complementan y refinan cada vez más con una evaluación genética del origen del sujeto.
Variación genética entre poblaciones.
En la vida cotidiana, dividimos a las personas en grupos según su apariencia o lenguaje de comunicación. La mayoría de los daneses se parecen más de lo que cada uno de ellos se parece a un italiano (
aquí hay una visualización genial con retratos promediados de diferentes nacionalidades). Los daneses e italianos están mucho más cerca uno del otro que cada uno de ellos, con los habitantes del África subsahariana: los fenotipos humanos se agrupan según el patrón geográfico. La distribución de genotipos tiene una estructura similar: los miembros de un grupo local, por regla general, tienen vínculos familiares más estrechos que los residentes de áreas remotas, y las poblaciones que viven en una región están más cerca que aquellas cuyos hábitats están separados por barreras geográficas (por ejemplo, cordilleras o agua). matriz).
Además, la diversidad genética de la población humana es menor que la de muchas especies biológicas. Esto se explica por el hecho de que la humanidad es una especie joven: los grupos individuales tenían relativamente poco tiempo para acumular diferencias. Dos personas seleccionadas al azar difieren entre sí por cada ~ 1000 nucleótidos, mientras que los dos chimpancés no coinciden una vez en ~ 500 "letras". Y, sin embargo, en total, hay alrededor de 3 millones de "puntos de diferencia" potenciales en el genoma humano. La mayoría de estas discrepancias, llamadas polimorfismos de un solo nucleótido (SNP), son neutrales o casi neutrales, pero algunas de ellas son responsables de las diferencias fenotípicas entre las personas.
La distribución de polimorfismos neutros (dado que no tienen un significado biológico, no están sujetos a una selección evolutiva direccional, son transportados por el viento de las migraciones) en la población mundial refleja la historia demográfica de nuestra especie. La evidencia genética y arqueológica indica que en los últimos 100,000 años el tamaño de la población humana ha crecido significativamente. La gente se estableció fuera de África, colonizando el resto del mundo. El proceso de reasentamiento afectó la distribución geográfica de los alelos de dos maneras: en primer lugar, el "efecto fundador" afectó: en la población de inmigrantes, por regla general, solo estaba representada una parte de las variantes genéticas de todo el conjunto de su diversidad en la población ancestral; en segundo lugar, tuvo lugar el llamado "cruce surtido", es decir Las parejas se formaron principalmente dentro de su grupo, lo que limitó la distribución de los polimorfismos de novo existentes y emergentes entre los individuos que habitan en diferentes áreas geográficas. Estos procesos llevaron a la acumulación gradual de diferencias genéticas.
En el contexto de los grupos de población, los marcadores genómicos comenzaron a estudiarse en los años 70 y 80, en los 90 comenzaron a usarse para identificar la población de una persona en particular. Los investigadores han demostrado repetidamente que los polimorfismos genéticos pueden aislar con éxito grupos de población y determinar la afiliación grupal de un individuo. Luego se demostró que las personas que viven en el mismo continente generalmente están más cerca unas de otras genéticamente que las personas de diferentes continentes. Inicialmente, en tales estudios, la información sobre el lugar de nacimiento, la raza, el grupo étnico se conocía desde el principio y se utilizaba junto con datos genéticos; Si los sujetos se distribuían a ciegas entre los grupos únicamente sobre la base de rasgos genéticos, la correspondencia entre el origen geográfico, el origen étnico y la estructura de la población era menos pronunciada. Como han demostrado otros estudios, el éxito dependía de los marcadores genéticos utilizados y su número (más es mejor), la elección correcta de las poblaciones de referencia y otros factores (
12 ).
En 2004, en los Estados Unidos, la definición genética de población se utilizó no solo en la investigación biomédica, sino también en investigaciones de delitos:
este artículo de Nature contiene una emocionante historia sobre cómo los agentes de policía, desesperados por encontrar un criminal, ordenaron una prueba de ADN de una empresa comercial, decidieron color de la piel del sospechoso y abrió el caso. Las sugerencias para el análisis del origen genético alcanzaron con éxito la ola de interés general en las personas en su propio pasado. "Roots mania", así llamado este hobby en un artículo en Time, dedicado a "La última obsesión de Estados Unidos": la investigación genealógica.
Los métodos genómicos son utilizados activamente por especialistas que estudian el origen y la evolución de los pueblos. Por ejemplo, en 2013, un equipo internacional de investigadores utilizó el análisis genético para refutar la hipótesis del origen de los judíos Ashkenazi de los jázaros (
13 ). El conjunto de datos genómicos utilizados por los autores es de dominio público: contiene más de 100 poblaciones mundiales. Proponemos simular un pequeño estudio con nosotros: determinar el lugar de los clientes de Genotek en esta muestra y, al mismo tiempo, comprender los detalles técnicos para determinar la población.
Propósito de la investigación
Identificar clientes Genotek entre poblaciones de referencia. Averigüe si hay representantes de judíos Ashkenazi en nuestra muestra. Demostrar los principios y métodos de análisis de la población de un individuo.
Objetivos de investigación
Procese los datos de genotipado de 722 sujetos utilizando el programa ADMIXTURE utilizando el conjunto de datos de Behar et al., 2013 como muestra de capacitación.
Materiales y métodos
El trabajo original de Behar et al., 2013 utilizó datos de 1.774 personas: entre ellos, representantes de 88 poblaciones no judías (de Arabia, Asia Central, Asia Oriental, Europa, Medio Oriente, África del Norte, Siberia, Asia del Sur y sub África sahariana) y 18 poblaciones judías. Los autores necesitaban un extenso conjunto de datos para determinar con precisión el lugar de los ashkenazes en el contexto de las poblaciones mundiales: la tarea consistía en presentar las tres regiones geográficas de las que este grupo podría venir hipotéticamente: Europa, Oriente Medio y Khazar Khaganate. Los autores enfatizaron la diferencia entre el enfoque de muestreo, que representa a las poblaciones modernas europeas, del Medio Oriente y judías, descendientes directos de poblaciones ancestrales, y muestras correspondientes al Khazar Kaganate, que dejó de existir hace aproximadamente 1000 años. El problema es que ninguna de las poblaciones existentes es el heredero directo del Khaganate. Los autores eligieron a los residentes del Cáucaso del Sur (abjasios, armenios, azerbaiyanos, georgianos), el Cáucaso del Norte (Adygs, Balkars, Chechens, Kabardins, Osetios y varias otras nacionalidades), Chuvash y Tatars como posibles representantes modernos de los jázaros.
Agregamos muestras de 722 personas de varias regiones de Rusia al conjunto de datos.
Para el análisis estadístico, utilizamos el programa ADMIXTURE, que nos permite estimar el origen más probable de un individuo en función de los datos sobre genotipos. Además, los autores del artículo en discusión utilizaron otros métodos estadísticos que dieron una respuesta similar a la pregunta planteada. Nos centraremos en ADMIXTURE, ya que es este algoritmo el que nos permite estimar el porcentaje de contribución de las poblaciones ancestrales a los genomas estudiados.
ADMIXTURE utiliza métodos de Monte Carlo en las cadenas de Markov (cadena de Markov Monte Carlo, MCMC). Aquí hay un
enlace a un artículo de los autores del algoritmo para aquellos que desean comprender con más detalle el lado matemático del proceso.
Veamos cómo funciona ADMIXTURE en el ejemplo de muestras y poblaciones de nuestro conjunto
En total, tenemos 2.496 muestras / individuos, cada uno de los cuales pertenece a una de las 106 poblaciones modernas. Sugerimos que las poblaciones modernas probablemente desciendan de un número relativamente pequeño de poblaciones ancestrales. Las "poblaciones ancestrales" en este análisis son algunos grupos genómicos antiguos, unidos por el principio de similitud genética. ADMIXTURE permite tanto suponer arbitrariamente suposiciones sobre el número de tales grupos en la muestra, como seleccionar el número óptimo que describa más correctamente la distribución real de los datos genómicos.
Habiendo recibido información sobre los genotipos y el número estimado de poblaciones "ancestrales" (K), ADMIXTURE construye un modelo que estima la contribución de cada una de las poblaciones "ancestrales" a cada muestra. Al interpretar los datos, tanto la composición cuantitativa del genoma (porcentaje de grupos) como la cualitativa son importantes: su presencia o ausencia en genomas específicos. Con base en estos datos, uno puede hacer suposiciones sobre los procesos evolutivos en una población, en particular, sobre la presencia o ausencia de "raíces" comunes en los grupos de población. Sin embargo, las conclusiones serán legítimas si el modelo que construimos es bueno: se selecciona el valor óptimo de K.
Seleccionamos el valor óptimo de K
¿Cómo determinar cuántas poblaciones “ancestrales” coinciden más con la verdadera para una muestra dada? Empíricamente!
ADMIXTURE es un programa inteligente: al construir un modelo de la estructura genética de las poblaciones basado en datos sobre los genotipos de los individuos (evaluando la contribución de cada uno de los grupos genómicos antiguos a cada uno de los genomas de la muestra) para un número dado de K, no se olvida de hacer una comparación con la realidad al final. Verifique qué tan bien la entrada es descrita por el modelo construido. Una medida de comparación es el "error", un valor que describe la falta de coincidencia entre el modelo y los datos reales. Cuanto mayor es el error, peor es cierto el supuesto del número de poblaciones ancestrales.
¿Cómo elegir el valor óptimo de K? Comenzamos el algoritmo ADMIXTURE en esta muestra, sustituyendo diferentes valores de K, y obtenemos para cada K su propio valor de error. Trazamos la dependencia de la magnitud del error en K. Aquí está el gráfico obtenido por los autores del artículo:
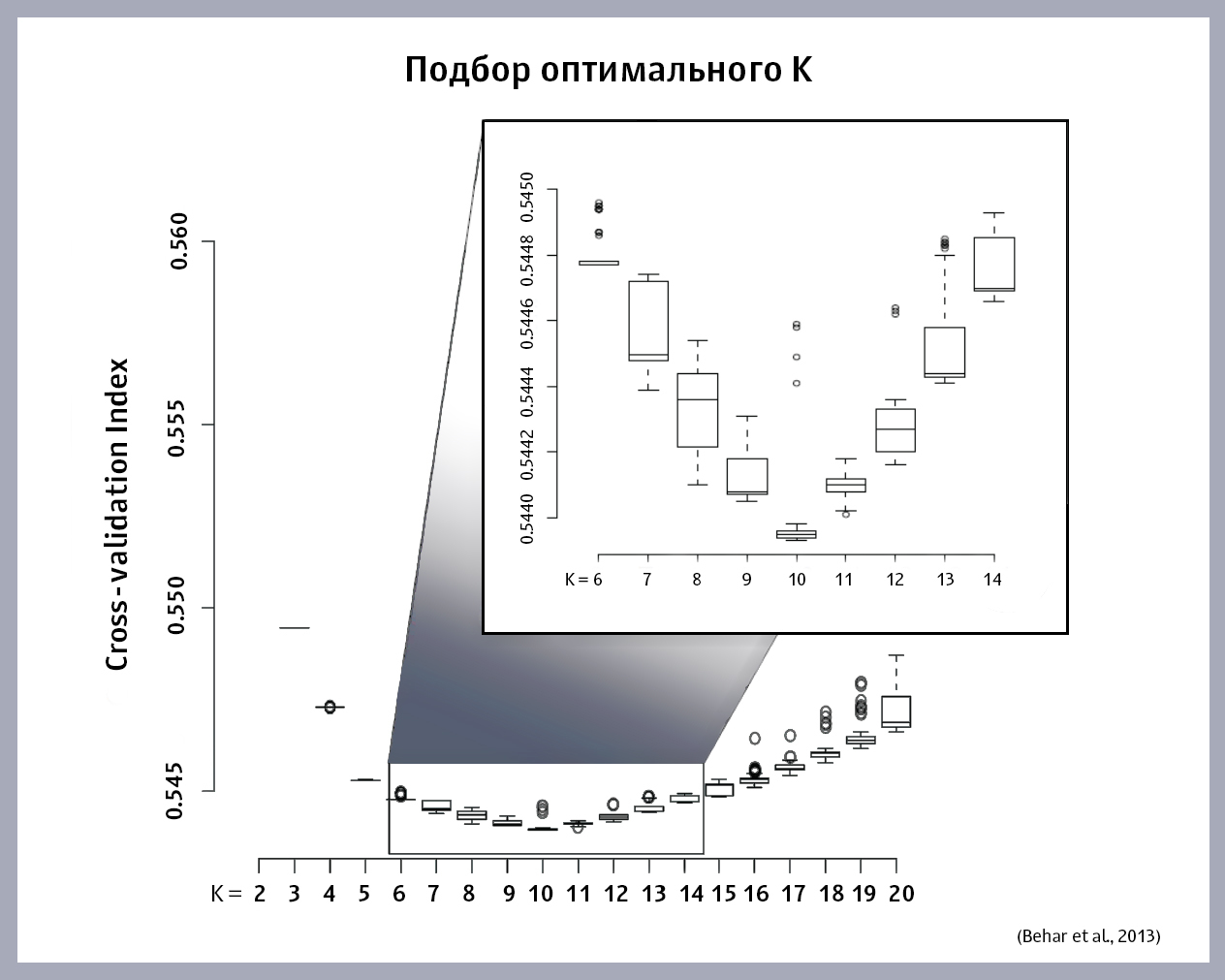
El valor óptimo de K está en el punto mínimo de la función. Si no se encuentra el mínimo en el gráfico (la función crece o disminuye constantemente), tendrá que construir modelos eligiendo nuevas K hasta que pueda encontrar la correcta.
Incluso con una K óptimamente seleccionada, la confiabilidad de los resultados del análisis depende de la exactitud de la muestra:
1. Las personas no deben estar relacionadas entre sí.
2. Los polimorfismos de un solo nucleótido (SNP) utilizados para genotipar deben distribuirse uniformemente sobre el genoma con una densidad suficientemente alta.
3. Los alelos SNP deben estar en enlace de equilibrio, es decir, la probabilidad de la presencia de un alelo dado en un individuo en particular debe depender solo de la frecuencia de este alelo en la población, y no de otros alelos en el genoma.
Como se puede ver en el gráfico, la K óptima para esta muestra fue de 10 poblaciones "ancestrales".
Resultados
ADMIXTURE visualiza los resultados del análisis de esta manera (en la figura solo se ve una parte de los datos):
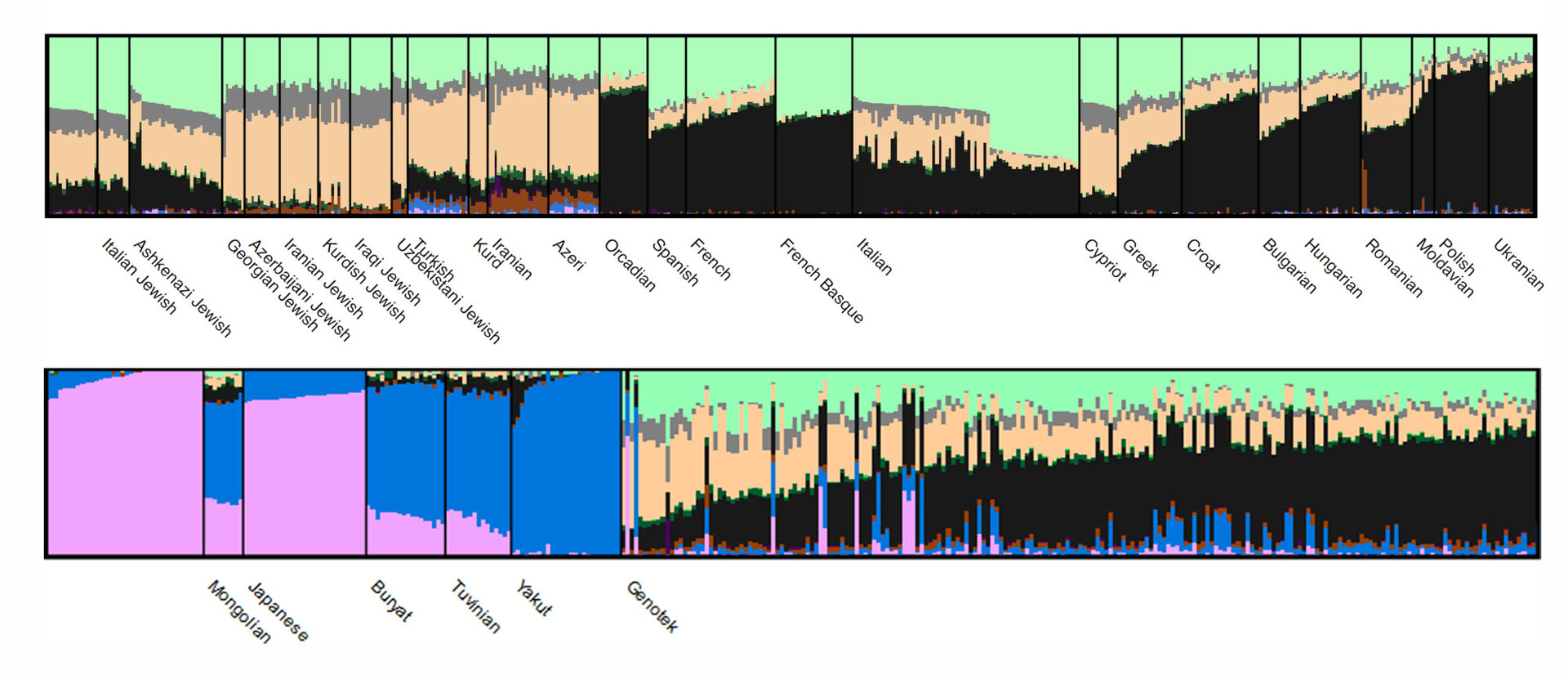
Cada grupo tiene su propio color, y las poblaciones difieren (o no difieren) en la proporción de grupos en el genoma.
Aquí se encuentra la versión interactiva de la imagen para un estudio detallado: mueva el mouse y desplácese para ver todas las poblaciones o para considerar algunos de los grupos con más detalle.
En general, dentro de la "población" de Genotek, se espera que la proporción de conglomerados corresponda al patrón característico de las poblaciones de origen de Europa del Este. Lo interesante comienza a nivel de muestras individuales:
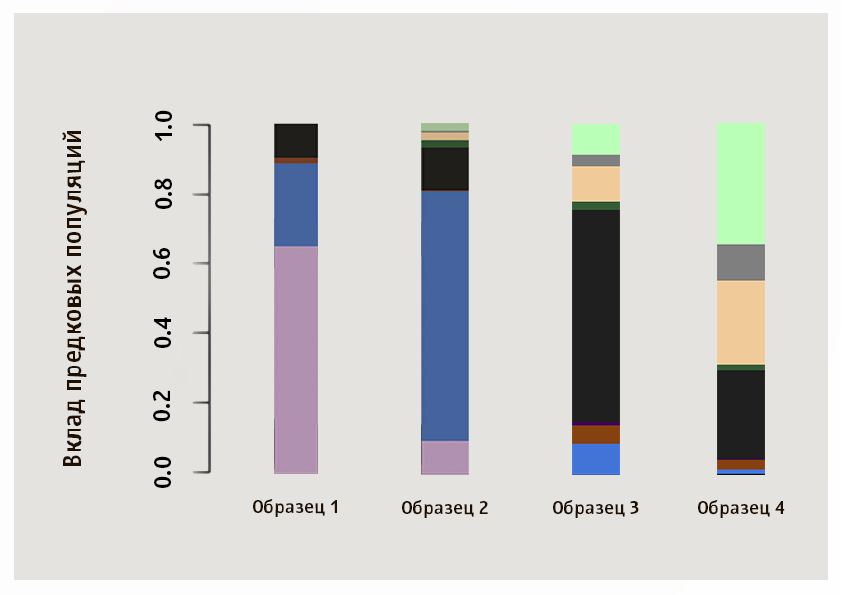
Aunque la población más cercana a la muestra dada se determina mediante valores numéricos, también se puede obtener mucha información mediante la comparación visual de patrones. Sugerimos que determine independientemente las poblaciones más cercanas para muestras de cuatro clientes Genotek a partir de la imagen.
La respuestaEn esta imagen, las muestras 1 y 2 son de origen asiático: el predominio del racimo rosado es típico para los japoneses y los Khan en nuestra muestra, azul para los Yakuts, la tercera muestra muestra la proporción de componentes típicos de rusos, bielorrusos, ucranianos y polacos, y el cuarto es típico Ashkenaz Judio.
De 722 muestras, encontramos 9 judíos Ashkenazi.
Conclusión
La afiliación a la población está lejos de ser el único factor que determina la autoidentificación étnica de una persona. Sin embargo, todavía es posible revelar una correlación entre los grupos étnicos y la estructura del genoma de sus representantes. Tal análisis se utiliza tanto para fines científicos y médicos, como para el estudio de sus propias raíces por parte de todos los interesados. Al mismo tiempo, es importante comprender que los modelos se mejoran constantemente, y los resultados obtenidos para una mayor precisión deben considerarse junto con otros datos, por ejemplo, el árbol genealógico familiar.
Los autores del artículo original no encontraron evidencia del origen jázaro de Ashkenazi. Las pruebas genéticas, por supuesto, "saben" cómo identificar a los judíos; sin embargo, uno no debe olvidar que "la judería" es, en primer lugar, un estado mental.
En un futuro cercano, se lanzará en Genotek la prueba de ADN de Genealogía actualizada con resultados extendidos: aumentaremos el número de poblaciones a cientos, agregaremos poblaciones judías. Actualizaremos la información en su cuenta personal para todos los que nos hayan pasado su material genético. Si aún no está genotipado, lo invitamos a
unirse .
Referencias
- Foster M., Sharp R. (2002). Raza, etnicidad y genómica: clasificaciones sociales como representantes de la heterogeneidad biológica. Genome Res.
- Collins FS, McKusick VA (2001). Implicaciones del Proyecto Genoma Humano para la ciencia médica. JAMA
- Nebert DW, Menon AG (2001) Farmacogenómica, etnia y genes de susceptibilidad. Farmacogenómica J.
- Olden K., Guthrie J. (2001). Genómica: implicaciones para la toxicología. Mutat Res.
- Yudell M., Roberts D., DeSalle R., Tishkoff S. (2016). Sacando la raza de la genética humana. Ciencia.
- Ogura, Y. y col. (2001) Una mutación de desplazamiento de marco en NOD2 asociada con la susceptibilidad a la enfermedad de Crohn. Naturaleza.
- Hugot, JP y col. (2001) Asociación de variantes repetidas ricas en leucina NOD2 con susceptibilidad a la enfermedad de Crohn. Naturaleza.
- Inoue, N. (2002). Falta de variantes comunes de NOD2 en pacientes japoneses con enfermedad de Crohn. Gastroenterología
- Martin, MP y otros (1998). Aceleración genética de la progresión del SIDA por una variante promotora de CCR5. Ciencia.
- González, E. y otros (1999). Efectos modificadores de la enfermedad por VIH-1 específicos de la raza asociados con los haplotipos CCR5. Proc. Natl Acad. Sci. USA
- Shi, Hong y col. (2009) La temperatura invernal y los rayos UV están estrechamente relacionados con los cambios genéticos en la vía supresora de tumores p53 en Asia oriental. American Journal of Human Genetics.
- Bamshad M., Wooding S., Salisbury B. et al. (2004) Deconstruyendo la relación entre genética y raza. Nat Rev Genet.
- Behar DM y col. (2013) No hay evidencia de datos de todo el genoma de origen jázaro para los judíos asquenazíes. Biología humana