 Puede decir todo lo que quiera que hay algo mal con la medicina en Rusia, pero nuestra protección al consumidor es una de las mejores del mundo.
Puede decir todo lo que quiera que hay algo mal con la medicina en Rusia, pero nuestra protección al consumidor es una de las mejores del mundo. En comparación con nuestros estándares europeos para dispositivos médicos, es solo un relleno.
En los últimos años, tenemos que demostrar cualquier efecto. Es decir, si encuentra una pomada de arrugas, pero no elimina las arrugas, puede quejarse a Rospotrebnadzor. Y en algún lugar lejano de la industria se puede escuchar el sonido de un frasco de vaselina que se abre. Si te sientes enfermo después de tomar un bálsamo curativo, también puedes quejarte. Y de nuevo, una persona, acudirán a alguien con un cheque.
Toda esta historia de requisitos de endurecimiento nos afectó seriamente hace un par de años cuando registramos Blefarogel. Pero déjenme decirles en detalle cuánto vaselina, lágrimas y mocos hemos gastado en este proceso.
4 círculos del infierno
La base para el registro de dispositivos médicos es el
Decreto No. 1416 .
Describe las reglas y el procedimiento para el registro. Para someterse a este procedimiento bastante complicado, se deben realizar las siguientes pruebas:
- Toxicológico (lo que demuestra que el producto es seguro en términos de materiales y sustancias utilizados en la fórmula).
- Técnico (que demuestra que el producto, en sus propiedades y características técnicas, cumple con los requisitos de TU y la documentación operativa del fabricante, así como el hecho de que el proceso de producción será uniforme y no será necesario verificar cada elemento individual del lote).
- Clínico (demostrando la efectividad y seguridad de resolver un problema médico).
- Y un examen de calidad, eficiencia y seguridad, donde la comisión recopila todos los datos sobre el producto en un montón y decide la posibilidad de su registro y lanzamiento al mercado.
Un punto importante:
por ley, un dispositivo médico no puede sanar. Es decir, si tiene un efecto terapéutico, entonces este medicamento es una categoría completamente diferente. El dispositivo médico puede ser algo auxiliar en el proceso. Por ejemplo, si se registra un tonómetro, se debe demostrar que mide la presión, que la manga en los puntos de contacto con la piel no causará alergias o diversas dermatitis, que la presión se medirá con una tolerancia dada durante toda la vida útil del dispositivo. En el caso de un pato, es necesario demostrar su hipoalergenicidad y seguridad para el paciente. En el caso de nuestros geles, lo principal es pasar por Roszdravnadzor, incluida la toxicología.
La toxicología solía ser relativamente simple. Hoy necesitamos comenzar con el análisis de componentes y sus interacciones. Lo importante es que esto concierne no solo al producto en sí, sino también al empaque. Anteriormente, solo verificaban el gel, y ahora, de acuerdo con los nuevos estándares, el gel y el empaque (frasco con tapa) de todos los envases y colores usados, así como su interacción. Esto se hace para que la sustancia de la botella no cambie las propiedades del gel, migrando a su composición.
Primero debe realizar una revisión de la literatura, es decir, encontrar información en forma de artículos y revisiones sobre análogos registrados que demuestren su efectividad y seguridad, encontrar prácticas sobre sustancias activas y demostrar que son seguras en la mezcla.
¿Por qué es la primera etapa de la revisión literaria? Permítame recordarle que no tenemos un medicamento, sino un dispositivo médico. No hay necesidad de probar la nueva fórmula. Más a menudo que no.
Se ve así: nos referimos a los artículos para su examen, es decir, encontramos revistas en los archivos, notariamos capturas de pantalla y copias para que la comisión en sí no mire más tarde.
Entonces necesitamos comentarios de la práctica clínica sobre análogos registrados. Es decir, debemos encontrar publicaciones (o tomar el análogo más cercano ya registrado una vez y verificar cómo resuelve este problema). Sobre la base de la información recopilada sobre análogos registrados, se elabora un acto de evaluación de los resultados de los ensayos clínicos de dispositivos médicos.
A continuación, con un paquete de documentos que confirma la finalización de todos los pasos indicados anteriormente, así como con otros documentos adicionales para el dispositivo médico, se nos envía a Roszdravnadzor para su registro.
¿Cómo va el proceso?
Primero, una solicitud y un dossier para el producto futuro se envían a Roszdravnadzor. La aplicación describe el propósito, la clase de riesgo, el alcance y otra información sobre el dispositivo médico, así como información sobre el fabricante. Además de la aplicación, se adjunta un conjunto de documentos: protocolos, certificados de todas las pruebas realizadas, documentación técnica (TU), documentación operativa (instrucciones de uso, etiquetas), imágenes fotográficas, un informe de análisis de riesgos, información sobre documentos reglamentarios, un certificado de prueba de calificación y contratos de arrendamiento y otros documentos del fabricante.
Es decir, en el momento de la presentación de la solicitud, es necesario completar todas las pruebas internas (en nuestra producción, en nuestro propio laboratorio se hacen más difíciles que en los laboratorios estatales, porque es elemental más rentable ir la primera vez y no encontrar revisiones de las partes más adelante en el proceso de ventas). Luego pase la toxicología a uno de los laboratorios con acreditación estatal.
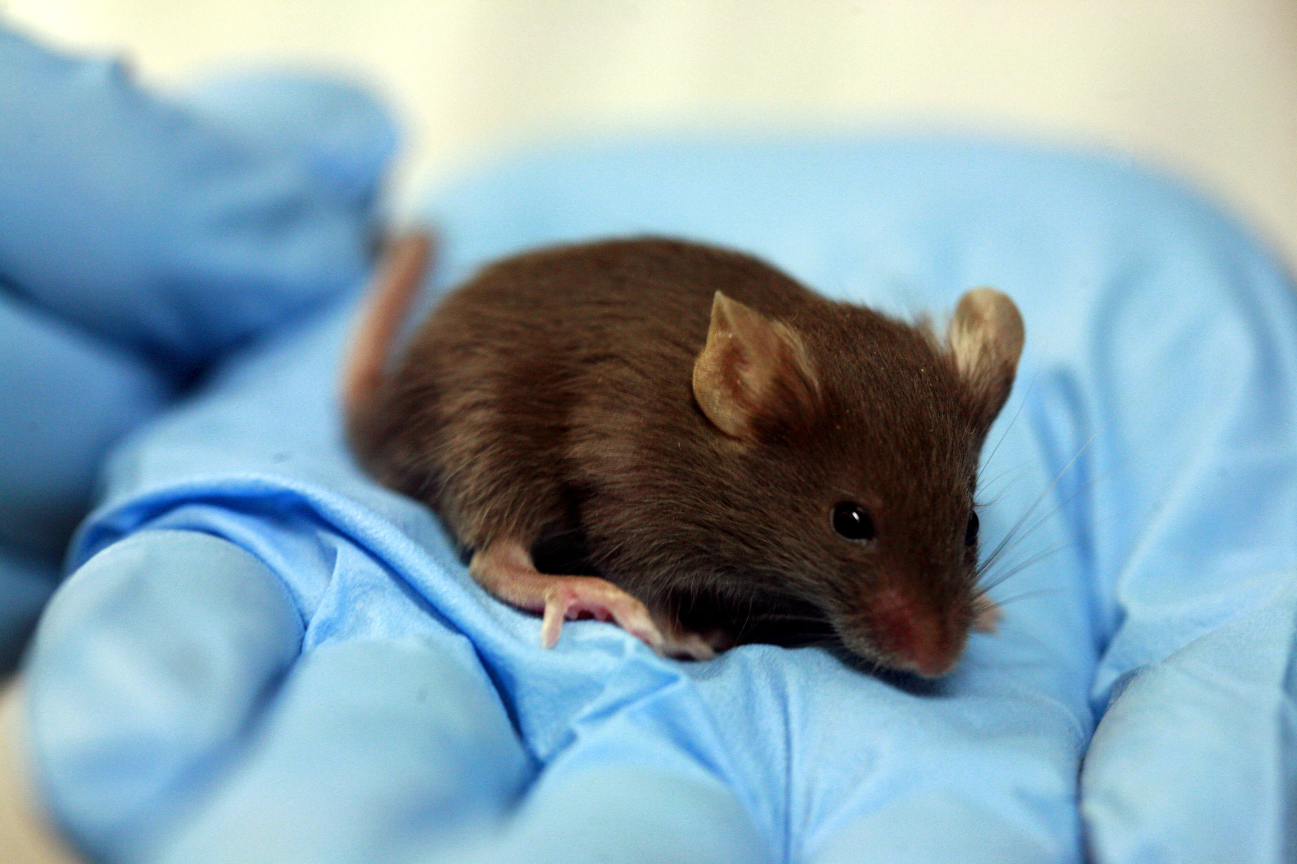
En nuestro caso, por ejemplo,
Blefarogel (un medio para la higiene de los párpados en la prevención
del síndrome del ojo seco ) es un gel que se aplica a los párpados y la piel del rostro. La toxicología fueron ratones. No sé qué hicieron exactamente con ellos, pero supongo que afeitaron dos áreas de cada animal, una untada con gel, la otra dejada o untada con algo neutro. Y miró al ratón. Afortunadamente, no empujaron dentro del mouse. (De acuerdo, casi no lo empujaron; el hecho es que en la hoja de datos de seguridad [o MSDS - hoja de datos de seguridad del material] de propilenglicol hay datos sobre la dosis letal cuando se toma por vía oral. Necesita 4 gramos por ratón [se rompe físicamente al mismo tiempo muy probablemente] Y después de todo, de alguna manera fijaron esta dosis ...) Pero volvamos a los productos para ratones más exitosos, que simplemente están manchados. Tienen vecinos que obtuvieron botellas limpias del producto, en las que vierten agua y se defienden. Luego se untan con esta agua de las botellas para verificar que el plástico no libera algo peligroso con el tiempo. O no: la metodología exacta no se ha revelado a los fabricantes, existe una alta probabilidad de que esta agua de la botella simplemente se analice químicamente y, incluso si se sospecha, se inyecte por vía intradérmica.
Sentimos lástima por los ratones, pero el enfoque es este: si no fuera por nuestras pruebas, no habrían nacido en absoluto. En general, no están atormentados allí (al menos, ya hemos entrado con un estudio listo sobre nosotros mismos). También sabemos que los asistentes de laboratorio tienen sentimientos cálidos por los ratones, comparable al amor de un aldeano por el pollo. Y sospechamos que al final de la investigación, el ratón no se elimina por completo, sino que se envía a rehabilitación antes de nuevas pruebas. Pero esto es solo una teoría: si hay alguien aquí que pueda contar sobre su destino, entonces sería maravilloso.
La misma solicitud debe ir acompañada de una investigación técnica. Específicamente, escriba TU (documento reglamentario para un dispositivo médico que establezca requisitos técnicos) y pruebe que el dispositivo médico le corresponde. Los expertos están de acuerdo con nuestras condiciones. Luego, se verifica la estabilidad de las muestras en serie y el cumplimiento de los expedientes de sus productos: color, olor, conductividad eléctrica, atenuación de la señal ultrasónica, etc., todo lo que se indica. En esta etapa, a menudo se requiere reformular una palabra en particular en la descripción del producto. Nuestro Mediagel tiene una viscosidad diferente: este es un certificado de registro, pero tres conjuntos de prueba son para viscosidad media, más líquido y gel más denso.
Llevamos a cabo las mismas verificaciones regulares para el cumplimiento de TU en nuestra propia producción. Solo una vez en toda la práctica hubo un caso cuando nos acercamos a los límites permitidos de la prueba, generalmente caminamos exactamente en el centro de las tolerancias entre los límites.
La clase de riesgo del producto está determinada por su propósito, método de uso, duración del uso, invasividad y otros criterios. Tenemos geles de ultrasonido, por ejemplo, la duración del contacto con la piel hasta una hora es la primera clase. Y en la segunda clase, por ejemplo, los espejos ginecológicos, diferentes cosas para los recién nacidos caen.
Hay una diferencia en los ensayos clínicos. Primera clase: los ensayos clínicos se pueden realizar de inmediato. La segunda clase y superior: la clínica solo después del permiso oficial de Roszdravnadzor. Antes de los ensayos clínicos, el equipo de desarrollo tiene derecho a realizar pruebas solo en ellos mismos. El objetivo es verificar el efecto principal. El objetivo de Beta es comprender los posibles efectos secundarios y varios casos interesantes, como "mientras toma otros medicamentos". Los ensayos clínicos comienzan cuando la seguridad del dispositivo médico está probada en teoría.
Observo que si no se trata de productos médicos, sino de cosméticos, se realizan estudios en voluntarios en clínicas acreditadas para investigaciones científicas de este tipo.
En la práctica, esto es lo siguiente: la comisión completa el análisis de los documentos (con la excepción de los dispositivos médicos de la primera clase de riesgo y los dispositivos médicos para el diagnóstico in vitro), luego envían una carta y dan 50 días hábiles para realizar estudios clínicos y proporcionarles los resultados. Para la mayoría de nuestros geles, toda la práctica clínica se obtuvo incluso antes de que se ajustaran los requisitos, por lo que no hubo dificultades. Pero ahora tenemos un pedido objetivo de oftalmólogos para el nuevo Blefarogel Forte: es como Blefarogel-2 con compuestos de azufre para demodex, solo para casos avanzados. Si Blefarogel-1 es "para que no me duelan los ojos mientras estoy sentado en la computadora", Blefarogel-2 es "para que no haya inflamación de la garrapata y no tenga nada que comer para siempre", entonces Blefarogel-Forte será "genocidio de garrapatas a toda costa". En el sentido de que aumentamos deliberadamente el riesgo de alergias al aumentar la proporción de sustancias activas, pero recombinamos la fórmula para que ayude en casos especialmente difíciles. Dado que esto se hace a pedido de los médicos que solo anticipan que necesitan dicha herramienta.
Fin de la epopeya.
A continuación, se verifica la integridad de los documentos en Roszdravnadzor: un consultor ejecutivo que simplemente verifica el paquete se destaca. Los documentos se escanean y se envían a una organización experta en el departamento de Roszdravnadzor. Como resultado, se establece una conclusión en Roszdrav sobre la base de la totalidad de todos los documentos. Lo convierte en un grupo de expertos. Si todo está bien y los documentos se corresponden entre sí, dan permiso para el registro estatal.
De ahora en adelante, nada se puede tocar en absoluto. Reemplazar la letra en la etiqueta o reemplazar el ícono automáticamente significa que necesita volver a registrarse. Nos pidieron de alguna manera cambiar la etiqueta de Mediagel-s a ruso-inglés. Esto significa volver a registrarse. Seis meses tardó en ingresar el texto en inglés.
Como el proceso es experto, se asocia en gran medida con interpretaciones específicas de normas bastante complejas. Esto significa que varias veces puede enviar documentos de acuerdo con el mismo esquema, y luego resulta que se necesitará una nueva hoja de papel. En general, este es un tipo de accidente: pueden terminar en cada reenvío. Al mismo tiempo, el antiguo registro estatal permanece y no desaparece, pero perdemos dinero para los juicios y pagamos un gran impuesto estatal incluso para el registro y los cambios. Y gastamos ratones estatales.
A continuación, debe esperar el pedido en papel. Cuando hay un pedido, el producto irá al registro, es decir, al
sitio en el que las farmacias analizan qué es qué. Solo a partir de este momento puedes hacer algo. La farmacia no acepta productos que no están en la RU del Ministerio de Salud, si no son cosméticos, por supuesto.
Solo en este momento puede comenzar la producción. Sin el número del Ministerio de Salud, no solo podemos vender, sino incluso producir en Rusia.
Todo, desde este momento, estamos haciendo el dispositivo médico en serie. Por experiencia: es muy importante contar con un laboratorio de control de calidad bien desarrollado, ya que cualquier queja significa una verificación exhaustiva en la fábrica (a juzgar por el comportamiento, no puede regresar sin un informe de al menos algunas violaciones) y detener los envíos. En este sentido, preferimos realizar pruebas de aceptación y pruebas periódicas por nuestra cuenta y cerrar los estudios de muestras pequeñas con las nuestras. Quiero decir que 10 voluntarios del estándar son pocos, pero 100 voluntarios ya son una muestra más o menos representativa. A la larga, una investigación más extensa siempre vale la pena, y para mí es un gran misterio por qué otros fabricantes no siempre hacen esto.
En el próximo post trataré de hablar sobre otra etapa importante de investigación: la prueba de desafío: esto es cuando las muestras están especialmente contaminadas con bacterias patógenas. Esta parte de la investigación nunca debe realizarse en ningún lugar cercano a los laboratorios de producción o investigación, por lo que se realizan las pruebas de desafío.