Introduccion
¿De qué trata este texto?
Si una persona escucha sobre una "simulación de la realidad", lo más probable es que se le ocurran varias obras de ciencia ficción (como Matrix, The Dark City o Zero Theorems), o juegos de computadora. En el caso de personas cuyas cabezas están obstruidas con un título de ingeniería, pueden
aparecer paquetes como
KOMPAS-3D AutoCAD, Solid Edge o NX. Una persona que está escuchando ciencia probablemente recordará cualquier
modelo de varios artilugios espaciales .
Pero hay un nivel más de Realidad, que resultará ser inmerecidamente olvidado: aquel en el que tiene lugar toda la química es el nivel de átomos y moléculas. También se puede simular con bastante éxito en una computadora. Dado que la mecánica cuántica está a cargo de todo en esta sección de Realidad, tales cálculos a menudo se llaman química cuántica. Y hablaremos sobre su conexión con la Realidad estudiada por métodos experimentales.
Este texto será sobre las cosas más elementales. Pero, la práctica de leer revistas científicas y escuchar varios informes muestra que esto debe recordarse constantemente.
El texto está diseñado para personas que entienden y / o están interesadas en cómo viven los átomos y las moléculas.Tomado de xkcd.comBreve antecedentesDio la casualidad de que un compañero, desafortunadamente, trabajaba en ciencia rusa, me invitó a dar una conferencia en su curso especial para 2 personas en una de las conocidas universidades físicas de Rusia. Pero, por una extraña coincidencia, fue transferida a una conferencia de estudiantes celebrada en paralelo ... Allí no despertó mucho interés entre los estudiantes, y lamentaba mucho el material, así que decidí enviarle un correo no deseado a Habr, tratando de convertir la conferencia educativa en un artículo de divulgación científica.
Métodos físicos para estudiar la vida de las moléculas.
Sabemos por los cursos de química y física de la escuela que todas las sustancias están compuestas de átomos, moléculas, iones o combinaciones de los mismos. Y parece que incluso sabemos qué tipo de vida viven. Pero esta información debe tener sus propias fuentes confiables (métodos de investigación), y realmente lo son.
Hay muchas, muchas formas de espiar la vida de los átomos. Aquellos que lo deseen, por ejemplo, pueden familiarizarse con algunos de ellos con más detalle en los libros de texto clásicos.
- Pentin Yu.A., Vilkov L.V. Métodos de investigación física en química. - M .: Mir, 2006,
- Drago R. Métodos físicos en química. - M .: Mir, 1981.
Pero, más o menos y con bastante facilidad, se destacan 3 grupos principales de métodos:
- métodos espectroscópicos
- métodos de difracción
- varios métodos de microscopía (no importa, translúcido o escaneado, para nosotros esto no es esencial ahora).
No se hablará de esto último, pero sus herramientas no son menos importantes que las dos primeras.
¿Por qué no se hablará de microscopía?(Simplemente no rehuyo la palabra en absoluto en microscopía)
Métodos espectroscópicos para el estudio de la materia.
Este poderoso grupo de métodos nos proporciona muchas, muchas cosas: desde la búsqueda y determinación de moléculas en el medio interestelar y en otros planetas hasta la verificación banal de explosivos en el aeropuerto.
El principio general de los métodos espectrales.
Cuando se habla de espectroscopía, generalmente se refiere al siguiente principio general de operación.
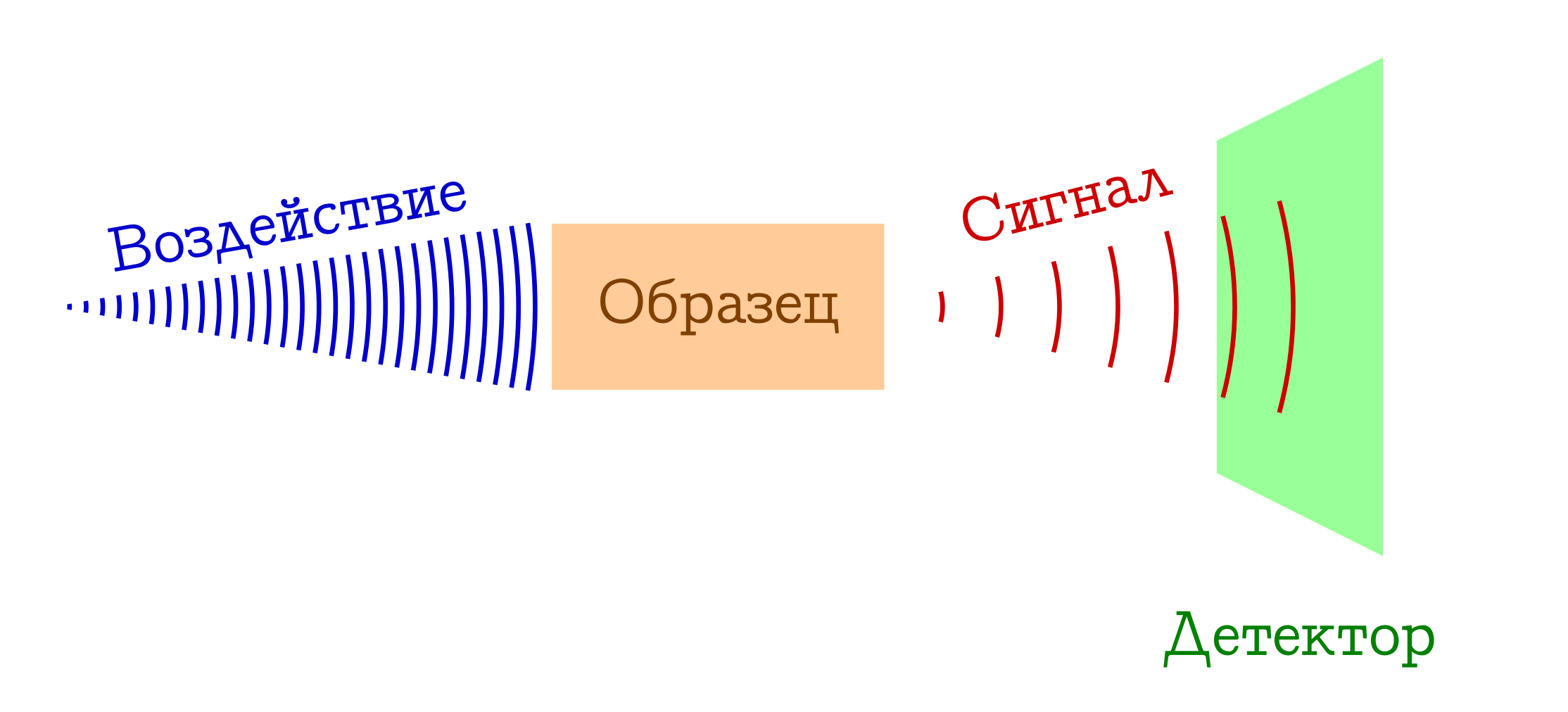
El esquema general de métodos espectrales para estudiar sustancias.
- Tenemos algo con lo que (por ejemplo, una bombilla / láser / luz solar) actuamos sobre la muestra que nos interesa. Muy a menudo, este es un estudio electromagnético, pero bien podría ser electrones (por ejemplo, en espectroscopía de masas con ionización por impacto de electrones) o un cóctel de todo lo posible e imposible del plasma (por ejemplo, en espectroscopía de llamas , tan querido por los escolares y estudiantes de la facultad de química). De una forma u otra, algo debe funcionar en nuestra muestra.
- Cuando se expone a una muestra, sucede algo que cambia su estado. Esto puede ser una transición a algún tipo de nivel excitado (en cualquier espectrofotometría o espectroscopía Raman), o incluso el colapso del sistema molecular (como en los espectros de masas o la espectroscopía de fotoelectrones ). Pero de alguna manera el patrón en algún momento debería ser diferente.
- ???
- BENEFICIO !!! Registramos una cierta señal (emitida o absorbida) con este cambio en la muestra a nivel molecular. Esto puede perderse fotones gastados en cambiar la muestra (luego tenemos espectroscopía de absorción), o viceversa, el exceso de fotones emitidos después de la excitación preliminar de la sustancia (espectroscopía de emisión), un cambio en la longitud de onda de los fotones iniciales como resultado de la interacción con la sustancia (espectroscopía Raman, más conocido en el extranjero como
Ramenovskaya Ramanova ), o fragmentos estúpidamente de las moléculas originales (como en espectros de masas o espectroscopía de fotoelectrones ). Hay muchas opciones, la esencia es la misma: ¡hay una señal!
Un ejemplo de tales métodos es un montón de letras diferentes: RMN, ESR, MW, THz, IR, UV / Vis, XRF, MS, PES, EXAFS, XANES, etc. etc.
Todos (o muchos de ellos) son familiares (o deberían ser familiares) para todos los químicos. Todos estos métodos son el arsenal estándar (lejos de ser incompleto) de un investigador respetuoso que se ocupa de sustancias.
Rangos espectrales y su relación con la vida de las moléculas.
 Tomado de xkcd.com
Tomado de xkcd.comDado que en la abrumadora mayoría de los casos la espectroscopía todavía está vinculada a la radiación electromagnética, es lógico vincular los rangos del espectro electromagnético a varios aspectos de la vida atómico-molecular. Después de todo, la frecuencia de las ondas electromagnéticas utilizadas en la espectroscopia es una especie de "reloj" que le permite detectar cuánto dura este o aquel proceso en los sistemas moleculares. Entonces, cambiando esta frecuencia, puede estudiar (e incluso actuar) en diferentes procesos moleculares.
Entonces
- Desde el punto de vista químico, no sucede nada interesante en el rango de longitud de onda súper larga, por lo que no puede recordarlo.
- Con la frecuencia de la radio y las microondas (e incluso la radiación infrarroja de onda larga, IR = IR), diferentes moléculas rotan en la fase gaseosa: grandes y pesadas, en la región de las ondas de radio (frecuencias más bajas), y pequeñas y livianas, en IR (frecuencias más altas).
- En el IR, sin embargo, (principalmente) ocurren varias vibraciones moleculares: todos los movimientos conformacionales y otros movimientos no obvios dentro de las moléculas están en el IR de longitud de onda larga, y las vibraciones de estiramiento (estiramiento - acortamiento de las longitudes de enlace químico) ocurren en la longitud de onda corta (hasta 4000 cm- 1 ).
- Bueno, entonces viene el lugar del espectro, donde viven varias transiciones electrónicas (hasta la región de γ-quanta). A frecuencias más bajas (visible, UV = UV y rayos X blandos), viven principalmente las transiciones asociadas con electrones de valencia.
¿Por qué lo vemos?Por cierto, es precisamente debido a las transiciones electrónicas que podemos ver: en nuestros ojos (en conos) hay estructuras que tienen una
retina en su composición. Cuando un fotón visible es absorbido por esta molécula, se rompe un doble enlace en él, lo que conduce a la isomerización cis-trans. Y es este cambio lo que percibimos como la señal primaria, que luego se transmite a nuestro cerebro.
Pero con el aumento de la energía de los fotones (es decir, con el aumento de la frecuencia, como recordamos de la fórmula de Planck E = h n u ) llegamos a capas cada vez más profundas de la estructura electrónica, hasta que descansamos en el rango de rayos X hasta las capas finales de 1s ( o, como se les llama rayos X, K ).
Entonces, al elegir la longitud de onda correcta de la radiación electromagnética, podemos observar con más detalle un proceso particular en las moléculas.
Métodos de difracción de sustancias
Ahora hablemos un poco sobre la difracción. El diagrama esquemático de tales experimentos también es simple.
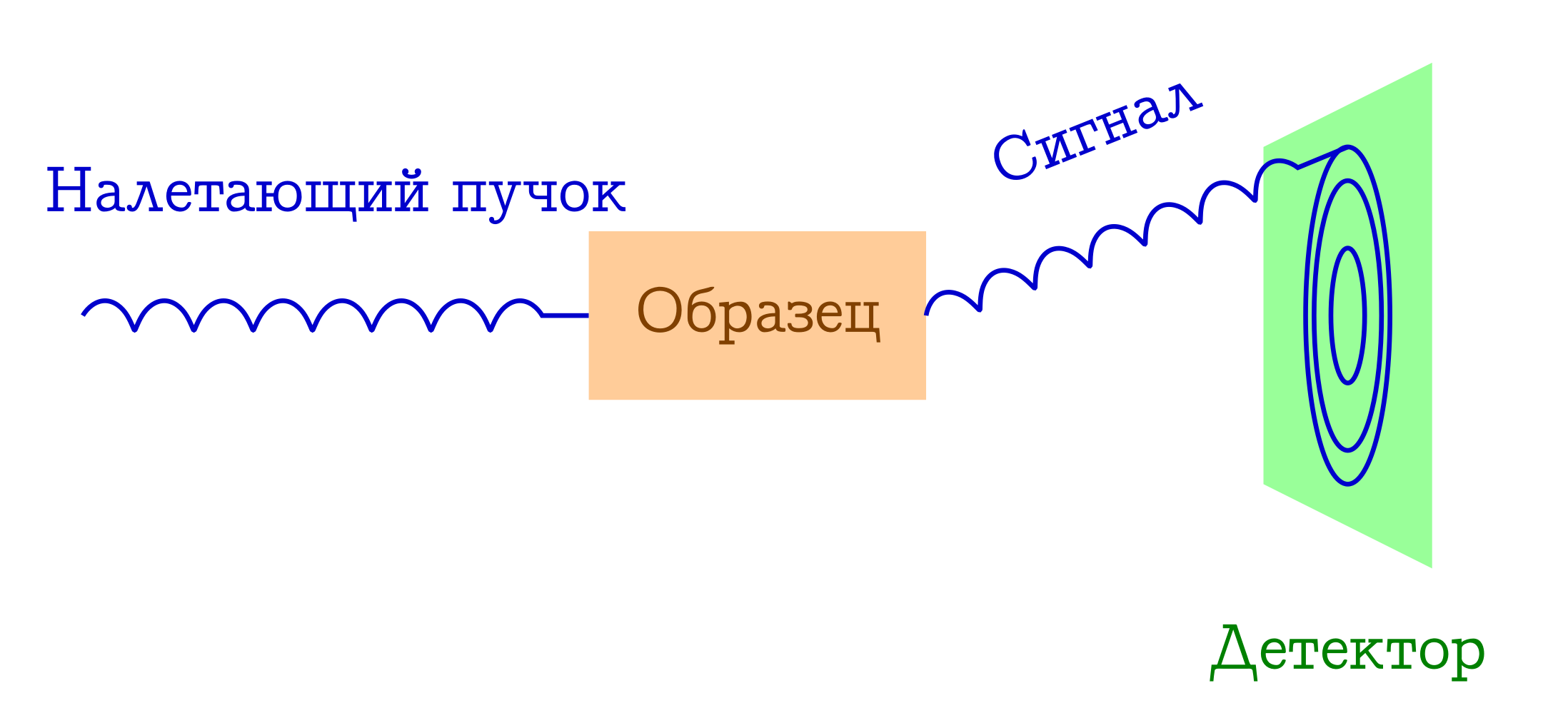
Esquema general de métodos de difracción para el estudio de sustancias.
- Un haz de algunas partículas vuela sobre la muestra. La mayoría de las veces son fotones de rayos X, electrones o neutrones.
- Estas partículas por diferentes mecanismos se dispersan elásticamente en los átomos de la muestra que nos interesa (es decir, sin cambiar la longitud de onda y la fase de la onda, simplemente cambian la dirección de su vuelo). No le sucede nada a la muestra de estas partículas incidentes: simplemente no tiene tiempo para reaccionar a ellas.
- Las distancias interatómicas sirven como una rejilla de difracción para el haz incidente, por lo tanto, como resultado, veremos una hermosa imagen de difracción en el detector.
Del último párrafo, surge la condición para la longitud de onda de las partículas incidentes (λ): debe ser del mismo orden o menor que el orden característico de las distancias interatómicas, por lo que λ típico para estos métodos es 1 - 0.01 Å.
Los principales tipos de errores al comparar experimentos y cálculos teóricos
Como resultado, tenemos una imagen muy interesante: en espectroscopía y difracción, observamos algún tipo de señal izquierda, que de alguna manera
indirectamente indica lo que realmente sucede en el sistema molecular.
La analogía con la cueva platónicaEsta pintura extrañamente recuerda el
mito de la cueva de Platón . Tenemos cierto mundo real de moléculas. Pero solo vemos sombras en la pared de la cueva (detector), que son una visualización incompleta de todas las cosas interesantes que suceden en este nivel de Realidad.
Pero, afortunadamente, a veces podemos calcular teóricamente la señal de interés para nosotros (como, por ejemplo, en espectroscopía de microondas, IR o UV / Vis), y a veces podemos extraer de la señal observada las cantidades de interés que están disponibles para el cálculo químico cuántico (por ejemplo, distancia entre átomos en una molécula, momento dipolar, etc.). Y aquí tenemos la posibilidad de que el experimento numérico y el real puedan unirse en la apasionante etapa de comparación entre ellos ... y aquí pueden ocurrir 4 tipos de errores como estándar.
Atencion El término "error" aquí no significa que el resultado de la comparación es obviamente incorrecto. Es solo que el terreno para la comparación se vuelve muy inestable y pantanoso, y un paso descuidado puede arruinar fácilmente todo el trabajo.
- Diferentes condiciones del experimento y / o cálculo (estado de agregación, temperatura, presión, etc.). De repente, podemos comenzar a comparar diferentes sistemas entre ellos, por alguna razón considerándolos iguales. Por ejemplo, es obvio que agregar una o cinco cucharaditas de azúcar a una taza de té conducirá al mismo sistema físico llamado "té con azúcar", pero las propiedades de este sistema serán muy diferentes. Y se puede medir fácilmente. Por ejemplo, con un termómetro (que mide la temperatura del té inmediatamente después de que se disuelve el azúcar) o con la lengua (uno de los llamados métodos de análisis organolépticos). Entonces, al comparar los sistemas resultantes entre sí (ya sea una verdadera taza de azúcar con té o su modelo de computadora), no debemos olvidar que las similitudes tienen sus límites, y que si reducimos el margen de error de "similitud", eventualmente encontraremos las diferencias.
- Diferentes significados físicos y / o matemáticos de los parámetros (el parámetro del significado físico en el sentido usual puede incluso no existir). Aquí, también, todo es simple: si comparamos 2 cantidades con un nombre similar, esto no significa que las cantidades tengan el mismo significado físico. Por ejemplo, la calificación de diputado entre toda la población de la ciudad vs. calificación solo entre abuelas. Tanto esta como esa calificación (sea lo que sea), estos números (o lo que sea) pueden incluso correlacionarse fuertemente entre sí, pero el significado de estos parámetros sigue siendo diferente, y esta diferencia se puede detectar.
- Errores "aleatorios" . Esto se refiere a algunos errores sistemáticos que el teórico del experimentador / simulador no conoce, o errores realmente aleatorios en el experimento / cálculo que no se pueden controlar y / o predecir. En principio, tales cosas pueden convertirse en el tema de investigación de varios efectos sistemáticos interesantes.
o simplemente una estimación de la relación S / N más útil ("señal a ruido"). - Y el último error estándar es el crecimiento de las manos del experimentador / calculador a partir del hueso pélvico , es decir, errores humanos comunes. No hay necesidad de investigar nada, solo verifique el trabajo o repita el experimento para encontrar y eliminar la jamba correspondiente.
No se puede decir nada más concreto sobre los dos últimos tipos de errores, sino sobre los dos primeros, y si toma un método de investigación específico, puede decir muchas cosas. Por lo tanto, nos concentraremos en ellos. El énfasis principal en este caso estará en las diferencias estructurales de las moléculas.
Error # 1. Diferencias en las propiedades moleculares en diferentes condiciones.
NaCl: cuando no hay errores
Por alguna razón, a nadie se le ocurre decir que el cristal único de cloruro de sodio (NaCl), que es una molécula enorme de iones Na
+ y Cl
- , y la molécula diatómica de NaCl, obtenida por evaporación de este cristal a temperaturas locas, tiene uno y el mismo Digamos la estructura.
E incluso si suponemos que al menos las distancias entre el cloro y el sodio (
r NaCl ) son las mismas aquí y allá, el experimento nos pondrá en su lugar:
¿Dónde nos equivocamos?De hecho, con tal comparación, permitimos la posibilidad del Error # 2, pero todo está bien aquí, si evaluamos los errores de tal comparación, serán del orden de 0.01 Å, que es significativamente menor que la diferencia de los parámetros comparados. Es decir Esto no es un error, sino un efecto real.
Cómo obtener la distancia entre el catión de sodio y el anión cloro en un cristal de sal usted mismoObtener la distancia entre los átomos en una molécula de NaCl diatómico a partir de datos experimentales tampoco es un procedimiento tan complicado. Pero el problema es solo que tal experimento es algo complicado. Por lo tanto, es más fácil usar una base de datos donde las distancias requeridas ya están dadas.
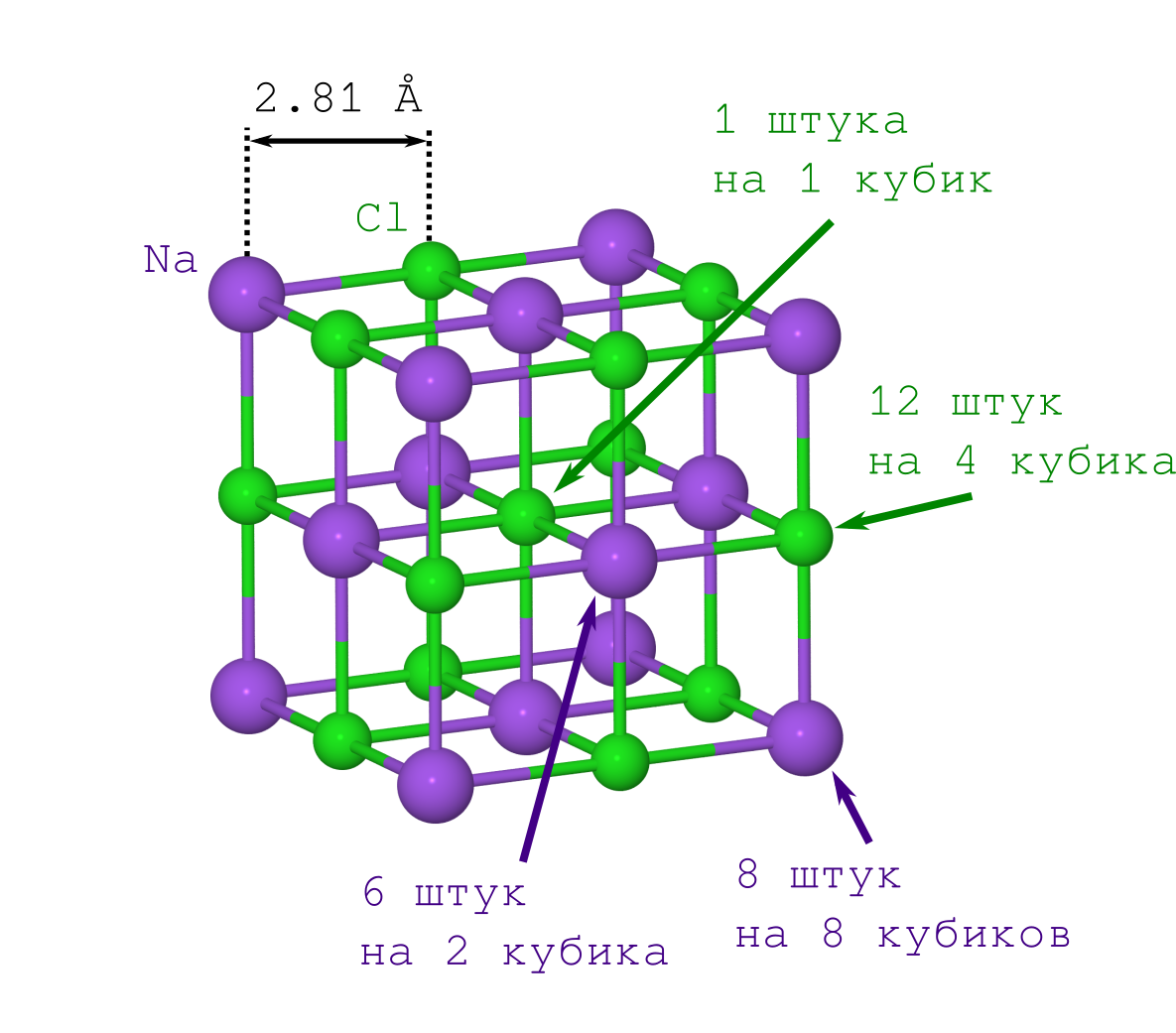
Pero para obtener la distancia entre los átomos en el cristal, solo la densidad de la sal cristalina de la tabla ρ = 2.165 g / cm
3 es suficiente, lo que puede
obtenerse fácilmente de Wikipedia y medirse en casa.
Para calcular la distancia necesitamos:
- la densidad del cristal de NaCl (es),
- conocimiento de la ubicación de los iones de este cristal.
Si hubiera hecho esto por primera vez (por ejemplo, a principios del siglo XX), tendría que atormentarse con el segundo punto. Pero esto ya lo saben las personas modernas: la red de NaCl tiene la forma de un cubo en el que los iones Na
+ y Cl
- se alternan entre sí (ver la imagen de arriba). Al multiplicar el fragmento indicado del cristal ("copiar y pegar" la pieza especificada y ajustarlo a la iteración anterior cara a cara), obtenemos un cristal de NaCl de cualquier tamaño y forma deseados (Minecraft).
Entonces, la densidad de este cubo debe ser la misma que la de todo el cristal. Dado que la densidad es
rho= fracmV (es decir, masa por volumen), resulta que conociendo la masa y la expresión geométrica del volumen, podemos calcular la distancia entre los átomos.
El volumen del cubo es obvio: la longitud de la costilla es el doble de la distancia Na - Cl (
L=2r mathrmNaCl ), lo que significa que el volumen deseado es
V=L3=8r mathrmNaCl3 .
La masa no es tan simple. La mayoría de nuestros átomos se encuentran en los vértices, bordes y caras del cubo, lo que significa que pertenecen simultáneamente a varios de estos cubos. Esto debe tenerse en cuenta en los cálculos.
Comencemos con los iones Na
+ . Tenemos solo 2 tipos de ellos (ver el patrón de red cristalina):
- los que están en los vértices del cubo (hay tantos como los vértices del cubo, es decir, 8, y están simultáneamente en 8 cubos, por lo que deberá dividir este número entre 8),
- los que yacen en las caras (hay 6 de ellos, y simultáneamente pertenecen a 2 cubos).
Como resultado, obtenemos que nuestro cubo contiene
8 cdot frac18+6 cdot frac12=4 ion de sodio
Ahora sobre Cl
- . También hay solo 2 tipos de ellos (ver el patrón de red cristalina):
- los que se encuentran en los bordes del cubo (hay 12 de ellos, y son propiedad conjunta de 4 cubos),
- ese Cl , que está en el centro del cubo, es uno y pertenece solo a nuestro cubo.
Por lo tanto, nuestro cubo contiene
12 cdot frac14+1 cdot frac11=4 ion de cloro
La composición del cristal, obviamente, corresponde a la fórmula química del NaCl, pero la masa de nuestro cubo es igual (no olvide que las masas de átomos en la tabla periódica se dan en
unidades de masa atómica ):
m=4 cdot( underbraceM mathrmNa23 textam+ underbraceM mathrmCl35.5 textamu)=234 textamu=234 cdot1.66 cdot10−24 textg=3.88 cdot10−22 textg\.
Ahora de la relación
rho= fracmV podemos hacer una ecuación para la longitud
r mathrmNaCl :
r mathrmNaCl3= left( fracm8 rho right) ,
que se resuelve fácilmente:
r mathrmNaCl= left( fracm8 rho right)1/3= left( frac3.88 cdot10−22 [ textg]8 cdot2.17 [ textg/ textcm3] right)1/3=2.82 cdot10−8 [ textcm]=2.82 [ textÅ] .
De los datos de la cristalografía de rayos X 2.81 Å (por ejemplo, de
Abrahams, SC; Bernstein, JL Precisión de un difractómetro automático. Medición de los factores de estructura del cloruro de sodio // Acta Crystallographica (1965) 18, 926-932 ) solo perdimos 0.01 Å, que es lo suficientemente genial
Alguien podría pensar que la diferencia de 0.45 Å es insignificante, pero este es casi el radio de Bohr (0.52 Å), que es igual a la distancia más probable del electrón, y para los estándares atómicos la diferencia es enorme.
Por qué el NaCl en forma de molécula atómica 2 difiere de un cristalAquí todo es muy simple. La red cristalina infinita crea la posibilidad de "salto" irreversible para 3s
1 electrón de sodio por átomo de cloro, ya que la diferencia de carga resultante se compensa con la interacción con los vecinos.
3s
1 ( ), ,
«» :
Na:Cl↔Na+Cl−
() (), .
,
±1 , , .
NaCl (2.36 Å),
d=q⋅rNaCl=9.0 [] donde
q≥0 (
+q ,
−q )
, « » 0.21, ..
d=0.21⋅9.0=1.9 [qe⋅Å] , :
q=drNaCl=1.92.36=0.8 . «» 0.2 NaCl NaCl .
Ferroceno
Vale la pena pasar de cristales iónicos a moleculares en los que las moléculas están densamente empaquetadas, por lo que de repente es posible comparar y sin ninguna reserva.Pero la diferencia no debe ser olvidada. E incluso hay un ejemplo clásico sobre este tema: una molécula de ferroceno .Esta es la conexión sándwich más simple. En él, un átomo de hierro neutro (como una chuleta) se encuentra entre dos anillos aromáticos de cinco miembros (bollos).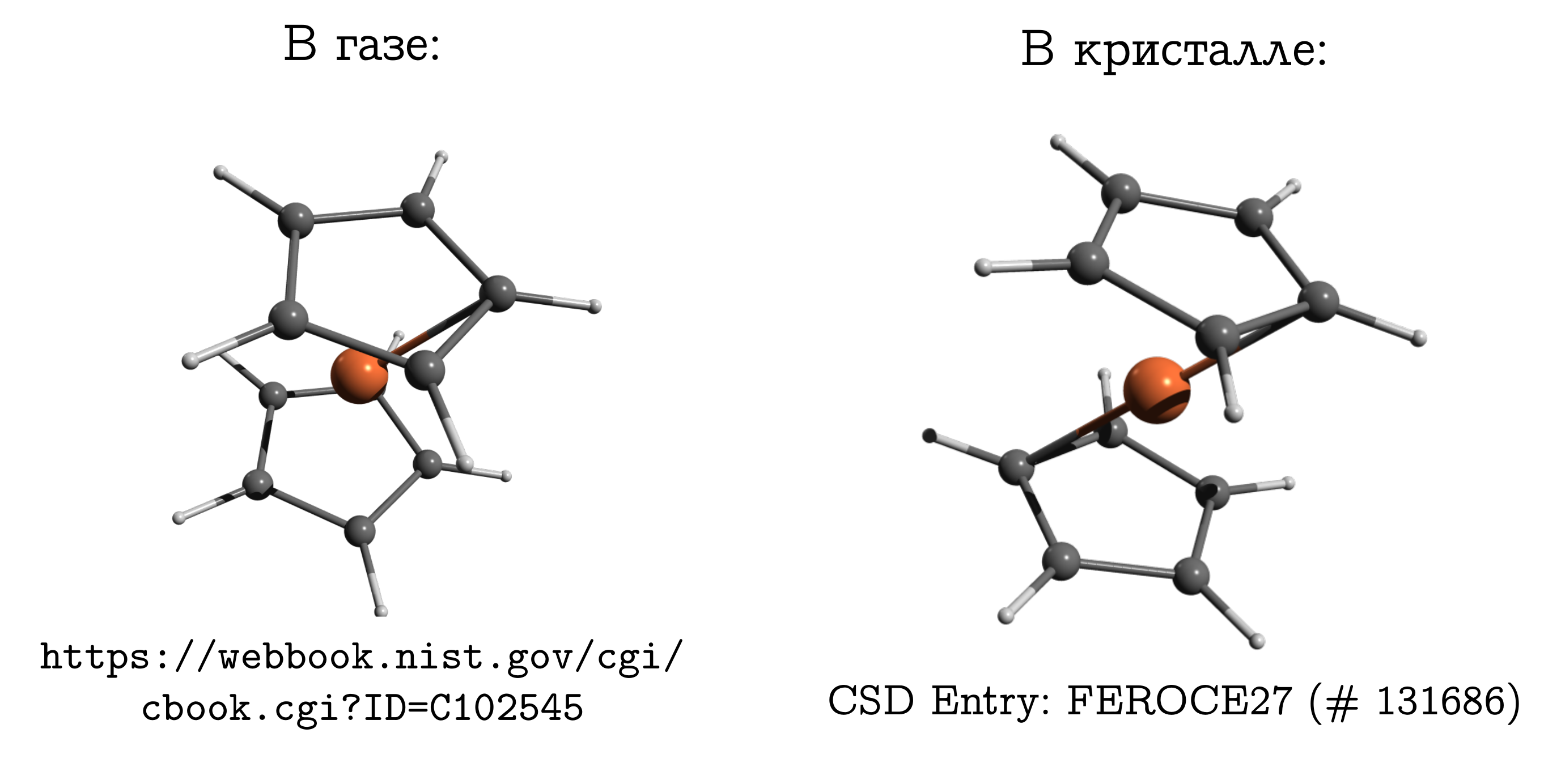 Esta molécula puede evaporarse con bastante facilidad y descubrir que la estructura más estable en la fase gaseosa es conformación obstruida En él, los carbonos e hidrógenos de los anillos superior e inferior están opuestos entre sí (ver la imagen de arriba), ya que en este caso las interacciones de dispersión son más fuertes. entre estas piezas de la molécula, y la dispersión siempre es beneficiosa.Si tomamos un cristal de ferroceno, resulta que las moléculas allí tienen una conformación estable diferente (que se llama inhibida para los hidrocarburos), en la que el hidrógeno y el carbono de un anillo están por encima / debajo del enlace C - C del otro. Hay interacciones de dispersión entre las moléculas, y un hecho similar, aparentemente inconveniente para una estructura de la molécula, surge del hecho de que es más fácil que las moléculas se unan solo en una forma incómoda, y este inconveniente personal se compensa con la interacción entre ellos.
Esta molécula puede evaporarse con bastante facilidad y descubrir que la estructura más estable en la fase gaseosa es conformación obstruida En él, los carbonos e hidrógenos de los anillos superior e inferior están opuestos entre sí (ver la imagen de arriba), ya que en este caso las interacciones de dispersión son más fuertes. entre estas piezas de la molécula, y la dispersión siempre es beneficiosa.Si tomamos un cristal de ferroceno, resulta que las moléculas allí tienen una conformación estable diferente (que se llama inhibida para los hidrocarburos), en la que el hidrógeno y el carbono de un anillo están por encima / debajo del enlace C - C del otro. Hay interacciones de dispersión entre las moléculas, y un hecho similar, aparentemente inconveniente para una estructura de la molécula, surge del hecho de que es más fácil que las moléculas se unan solo en una forma incómoda, y este inconveniente personal se compensa con la interacción entre ellos.¿Por qué el ferroceno es tan diferente del etano?Una persona familiarizada con la química generalmente tiene que dominarse para recordar la estructura del ferroceno en un gas. Después de todo, él tiene recuerdos de etano (C
2 H
6 ), en el que se inhibe la conformación más estable (cuando los hidrógenos de una pieza de CH
3 se encuentran "entre" los hidrógenos de otro CH
3 ), porque en esta posición, se minimiza la repulsión interatómica entre las capas electrónicas de los hidrógenos.

Adaptado de
www.chem.msu.su/rus/teaching/stereoY aquí toda la diferencia está en la distancia. La forma estándar del potencial de las interacciones de dispersión es el potencial de Lennard-Jones (esto, por cierto, es uno, no dos hombres):
V mathrmLJ(r)= fracAr12− fracBr6
En él, el primer término se toma de la repulsión interatómica, y el segundo de la atracción interatómica que surge de las fluctuaciones de la densidad electrónica. En general, este potencial se parece a esto:
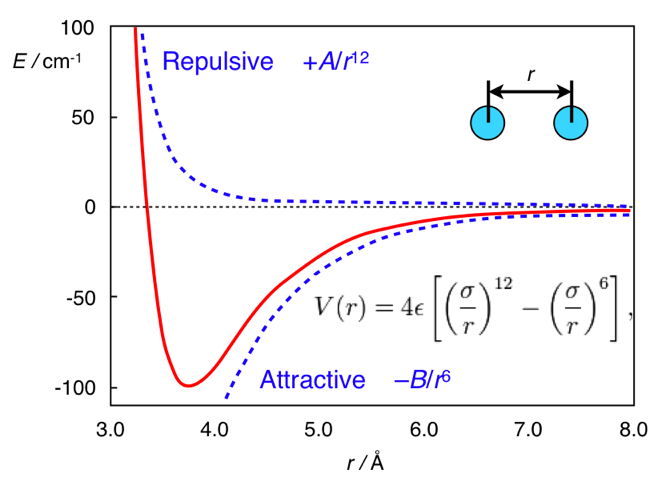
El potencial de Lennard-Jones. Adaptado de
chemistry.stackexchange.com/questions/34214/physical-significance-of-double-well-potential-in-quantum-bondingY en el caso del etano, los átomos de hidrógeno están demasiado cerca el uno del otro, por lo que están (en relación con su mínimo) en el lado izquierdo de la curva, y se caracterizan por la repulsión. En el caso del ferroceno, entre los anillos hay una capa de tamaño no enfermizo (átomo de hierro), por lo que los anillos están lo suficientemente lejos como para no sentir repulsión interatómica. Y entonces están en la parte correcta (atractiva) del potencial.
Histamina
En el caso del ferroceno, vimos el llamado diferencias conformacionales: la molécula permaneció igual (es decir, no se rompieron ni formaron enlaces químicos), y su forma cambió ligeramente.
Pero las diferencias pueden ser aún más fuertes, por ejemplo, si el llamado
Transformaciones tautoméricas . La tautomerización es una clase de reacciones químicas que ocurren tan fácil y rápidamente que, como resultado, podemos tener simultáneamente varios isómeros de una molécula, pasando fácilmente entre sí. Estos isómeros se llaman tautómeros.
Un ejemplo estándar de esto: tautomerismo ceto-enólico en cetonas:

Muy a menudo, como en este ejemplo, el tautomerismo se asocia con el salto de un protón de un lugar cálido a otro. Y estas reacciones están ligadas al
efecto túnel , al cual el hidrógeno, como el más ligero de los átomos, es más susceptible.
Dichas transformaciones químicas son características de muchas moléculas biológicas, por ejemplo, las
bases nitrogenadas que forman el ADN o los
azúcares .
Pero al pasar de un sistema a otro, las constantes de equilibrio de tales reacciones a menudo cambian, por lo que en diferentes fases podemos observar diferentes composiciones tautoméricas.
Un ejemplo de esto es la molécula de histamina (ver figura a continuación).
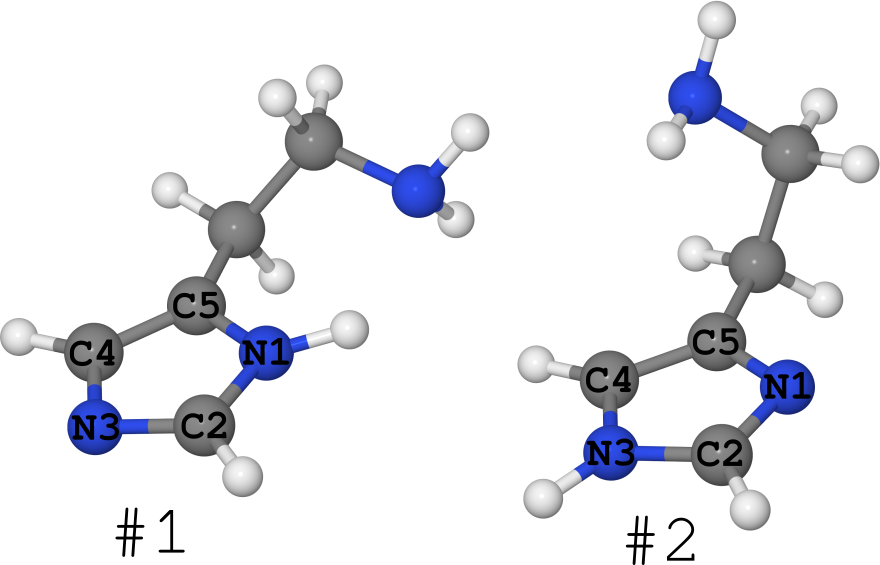
Existe en forma de 2 tautómeros (generalmente no callo sobre el número de conformadores, hay muchos de ellos):
- # 1, donde el hidrógeno se encuentra en el nitrógeno N1,
- # 2, donde el hidrógeno se encuentra en el nitrógeno N3.
Dio la casualidad de que para esta molécula se conocen sus estructuras en diferentes fases.
- En el cristal, está completamente "congelado" en forma de # 1. (vea el artículo del DOI: 10.1021 / ja00796a011 y la estructura en el Cambridge Structures Bank con el nombre "HISTAN" y / o el número 1176642)
- En soluciones acuosas, esta molécula existe en ambas formas, y el tautómero n. ° 2 es notablemente más grande ( DOI: 10.1021 / ja027103x ).
- En el gas, la histamina existe igualmente en la forma # 1 y en la forma # 2 ( DOI: 10.1021 / ja980560m ).
Es decir diferentes fases contienen diferentes números de diferentes moléculas, lo que significa que son diferentes sistemas.
Error Conclusión # 1

La principal conclusión a extraer de los ejemplos anteriores es la siguiente:
Al comparar cálculos en una fase con un experimento en otra, uno debe estar preparado para las diferencias sistemáticas.
Esto no significa que no sea necesario comparar: es necesario comparar, pero solo es necesario ser más crítico con las diferencias y / o coincidencias encontradas, y evaluar tales efectos si es posible.
Error # 2. Parámetros moleculares del "zoológico".
El segundo error se describe brevemente de la siguiente manera: si los parámetros se llaman de manera similar, pero no idénticamente, estos son parámetros diferentes.
Para comprender cuál es la fuente de este desacuerdo entre la teoría y el experimento, habrá que analizar con más detalle tanto los métodos experimentales estándar que se utilizan para obtener parámetros moleculares como los modelos que calculan cantidades similares exclusivamente a partir de la teoría.
Y aquí volveremos a hablar solo de estructuras.
Cómo obtener estructuras moleculares experimentales
Para limitarnos de alguna manera, solo hablaremos sobre métodos para estudiar la estructura de moléculas individuales, es decir sobre la fase gaseosa.
Tenemos dos fuentes principales de dicha información:
- difracción de electrones gaseosos,
- espectroscopía de microondas.
Nos detendremos en cada uno de estos métodos con más detalle.
Difracción de electrones gaseosos
El método es bastante antiguo, se origina en los años 30 del siglo XX, cuando los científicos alemanes Mark y Wirl realizaron los primeros experimentos sobre la difracción de electrones por gas.
Pocas personas lo saben, pero este método de investigación está involucrado en recibir tres Premios Nobel de química.
3 nobles con entrada electrográfica- Peter Debye en 1936 recibió su premio con la redacción:
"[por su trabajo en] la estructura molecular a través de sus investigaciones sobre los momentos dipolares y la difracción de rayos X y electrones en gases "
Esta es la única mención explícita de la difracción de electrones gaseosos en los méritos del laureado, y no sin razón. La ecuación básica de difracción de electrones para la intensidad de dispersión molecular se llama Debye.
de hecho, la ecuación de DebyeIij(s)=gij frac sin(srij)rij
Aqui
Iij denota la intensidad de dispersión de electrones (o rayos X u otras partículas) por un par de átomos
i- ésimo y
j- ésimo a una distancia
rij aparte
s= frac2 pi lambda sin left( frac theta2 right) ¿Es la coordenada de dispersión asociada con el ángulo de dispersión?
theta y longitud de onda de partícula
lambda y
g - la capacidad de este par de átomos para dispersar partículas difractantes.
Y a pesar de que se recuerda algo sobre esta maravillosa física (el modelo de soluciones iónicas , su modelo para calcular la capacidad calorífica de los cristales ), pero no la difracción de electrones, recibió el premio científico principal (en particular) por ello.
- Linus Pauling en 1954. Sí, el que recibió 2 Premios Nobel personales,
e incluso le dio a todo el mundo vitamina C , Gran Pauling. Cuando trabajaba en Kaltekh, en particular, se dedicaba a la difracción de electrones gaseosos (véase, por ejemplo, DOI: 10.1021 / ja01873a047 ). Y, por supuesto, el conocimiento de la química estructural de las moléculas libres lo ayudó a crear la famosa teoría del enlace químico (pero no minimicemos su gran fondo cristalográfico aquí). - Odd Hassel, Laureado 1969. Recibió su 1/2 Premio Nobel por el descubrimiento del equilibrio conformacional. Lo hizo sobre la base de un estudio de difracción de electrones de ciclohexano. Esta molécula existe en forma de dos conformaciones: una silla (silla) y un baño (en la tradición inglesa: un bote, un bote).

Desde aquí: www.shapeways.com/product/N5FE298DS/cyclohexane-2-molecules-boat-and-chair-form
Estas opciones para la disposición de los átomos se convierten rápidamente entre sí, pero en ese momento no lo sabían, y creían que solo una de las estructuras debería realizarse. Solo la señal de difracción de electrones no quería ser descrita por ninguna de estas estructuras, y solo una combinación de señales de ambas conformaciones podría explicar el patrón de difracción observado (se puede encontrar más sobre esto en el libro de I. Khargittai "Frank Science. Conversations with Famous Chemists").
El esquema del método en sí es muy simple (vea la imagen a continuación).
La cosa sucede en el vacío.
- Los electrones rápidos son eliminados continuamente del cátodo, que se aceleran en el campo anódico a energías de 40-60 keV.
- Los electrones suficientemente dispersos (pero rápidos) son enfocados por una lente magnética, luego de lo cual se convierten en un haz estrecho.
- Se instala una cámara con una sustancia perpendicular a la viga. La muestra se calienta a ebullición y el vapor resultante entra en contacto con el haz de electrones.
- Los electrones se dispersan con éxito por las moléculas, y silenciosamente vuelan más lejos, donde caen sobre la película.
- Por lo general, delante de la película pone el llamado. dispositivo sectorial. Esta es una pantalla giratoria muy rápida de una forma inusual. El hecho es que el electrón tiene la probabilidad de desviarse de su dirección original (por un gran ángulo de dispersión t h e t a ), cae muy rápido. Por lo tanto, para suavizar esta disminución de intensidad, el sector eclipsa uniformemente la parte central de la película, dejando abierta la parte más alejada. El resultado es una imagen más uniformemente iluminada.
- La trampa del rayo atrapa los electrones que no están dispersos (y hay muchos de ellos).
- Bueno, para que las moléculas no vuelen por todo el dispositivo y lo ensucien, se congelan en una trampa fría enfriada por nitrógeno líquido.
El resultado es el mismo patrón de difracción de anillos concéntricos descrito por la ecuación de Debye (esto es una señal). Varios parámetros moleculares pueden extraerse directamente de él.
¿Dónde puedo encontrar laboratorios de electrones de gas?No quedan muchos.
Pero en Rusia hay dos: Moscú (en el Departamento de Química de la Universidad Estatal de Moscú) y en la Universidad Química-Tecnológica de Ivanovo.
Espectroscopía de microondas
Este método de estudio de moléculas es más conocido, por lo que hablaré un poco más brevemente, usando la modificación más moderna como ejemplo: un espectrómetro de transformada de Fourier (como en ruso, en pocas palabras, espectroscopía de microondas transformada de Fourier).
El diseño aquí ya es más complicado, ya que requiere un montón de componentes electrónicos diferentes (amplificadores, moduladores de frecuencia, etc.). Omitiremos todo esto y hablaremos solo sobre lo que está sucediendo dentro de la cámara de vacío.
- Frente a frente hay dos antenas de bocina (como la que abrió el estudio de relicción ). Uno de ellos sirve como transmisor, y el segundo es un receptor.
- Perpendicular a estas antenas hay una válvula que lanza la muestra. Muy a menudo, se lanza en forma de vapor junto con un determinado gas portador (generalmente gases inertes) en el modo de expansión adiabática. Bajo tales condiciones, las moléculas se enfrían rápidamente a temperaturas cercanas a 0 K, lo que simplifica enormemente el espectro, haciéndolo más susceptible a la interpretación.
- Cuando las moléculas llenan toda la cámara, la antena transmisora las irradia con una señal lineal de frecuencia modulada. En la representación de frecuencia, esto corresponde a la suma de todas las frecuencias en un cierto rango.
- Algunas moléculas absorben esta radiación a diferentes frecuencias transmitidas y entran en un estado excitado. Pero, después de un tiempo, retroceden y comienzan a irradiar lo que han atrapado durante el impulso de la antena transmisora. Esta parada se ve como una señal de oscilación decreciente ( decadencia de inducción libre ). La segunda antena también lo registra. Luego, después de la transformada de Fourier de registrar esta señal en el tiempo, se obtiene el espectro de frecuencia habitual.
A diferencia de la difracción de electrones, que no era importante qué tipo de moléculas considerar, en una espectroscopía de microondas una molécula debe tener un momento dipolar constante (en casos raros, un momento dipolar magnético también es adecuado, esto es típico para radicales, como una molécula de O
2 ). La señal aquí es "intensidad de emisión vs. frecuencia ". Las constantes rotacionales se extraen de estos espectros a través de algunos modelos, de los cuales se extrae la estructura molecular.
¡Bienvenido al zoológico de parámetros moleculares!
Ahora es el momento de ver qué parámetros geométricos podemos obtener de varios experimentos. De hecho, cada uno de los tipos de cantidades indica qué tipo de modelo se utilizó para ajustar la señal experimental (con mayor frecuencia por el método de mínimos cuadrados). La mayoría de estos parámetros se pueden encontrar en la revisión Kuchitsu K., Cyvin SJ // En: Molecular Structure and Vibrations / Cyvin SJ (Ed.) - Amsterdam: Elsevier, 1972.- Ch.12. - P.183-211.
Comencemos nuevamente con la electronografía.
- rg= langler rangleT estructura Esto es solo un conjunto de valores promedio de distancias interatómicas a una temperatura dada.
- ra= langler−1 rangle−1T este valor es similar rg , pero es algo más natural para describir el patrón de difracción.
- r alpha=rh,0=? . Este valor no tiene un significado físico claro y está completamente vinculado al modelo interpretativo. De hecho, esto es lo que se observa en la cristalografía.
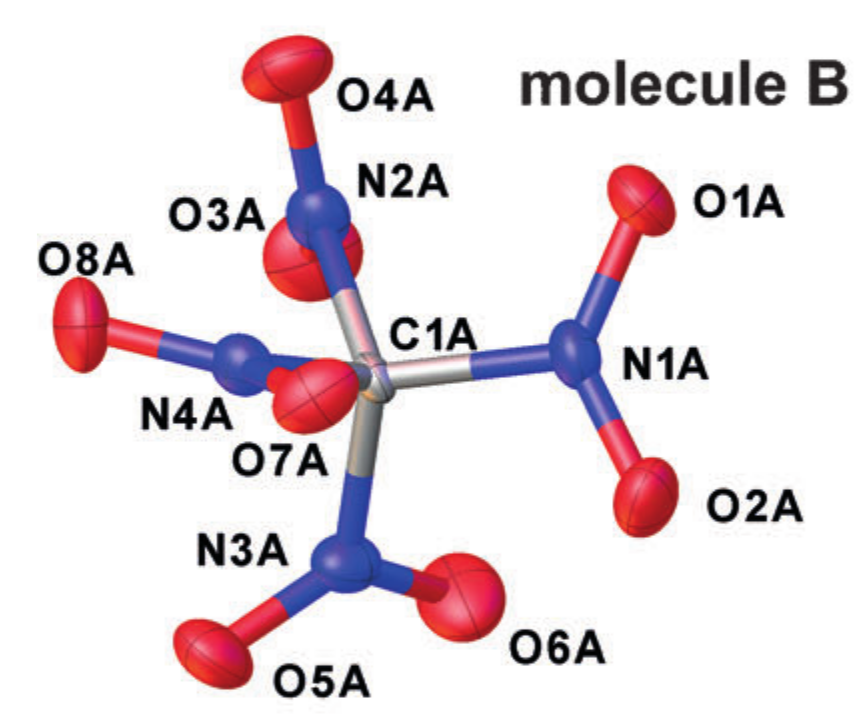
Ejemplo cristalográfico r alpha estructuras (tetranitrometano en el cristal). Adaptado de DOI: 10.1002 / anie.201704396
Cada átomo se aproxima mediante un elipsoide que describe su movimiento vibratorio, y las distancias entre los centros de las elipses resultantes se toman como distancias interatómicas. Pero, tal simplificación de la naturaleza del movimiento de los átomos corresponde a la introducción de la aproximación armónica del oscilador para las oscilaciones, y no siempre funciona bien.
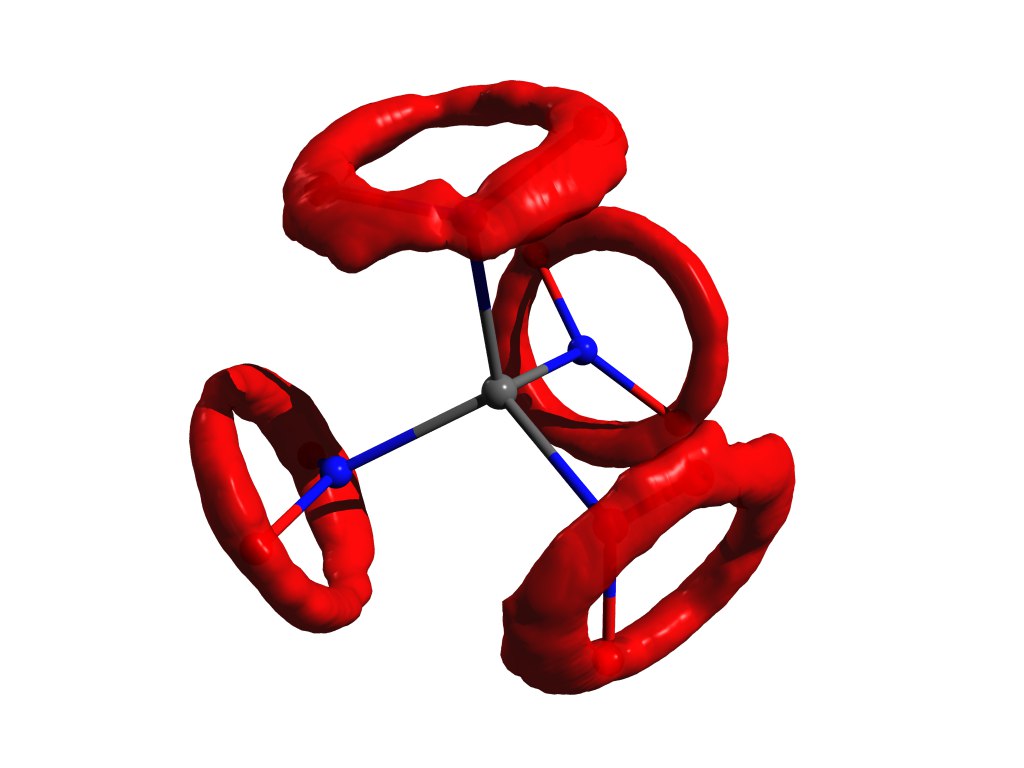
Un ejemplo de la distribución de átomos cuando la aproximación del oscilador armónico no funciona. El mismo tetranitrometano, pero en el gas.
- rh1=??? . Este
HEX milagro-yudo pez ballena no se describe en pocas palabras, tiene una relación muy débil con la realidad, pero tiene propiedades maravillosas: debe ser geométricamente consistente (ver más abajo) y puede calcularse fácilmente. Debido a esto, ganó su gran popularidad en la comunidad electronográfica.
En la espectroscopía de microondas, hay un poco menos de variantes estructurales.
- Lo más comprensible físicamente es rn= langlen| hatr|n rangle estructura De hecho, esta es la geometría promediada sobre un cierto estado vibratorio de la molécula ( |n rangle ) Como trabajan con mayor frecuencia con moléculas frías, generalmente observan una estructura específica de esta clase: r0 es decir La geometría de la molécula en el estado vibratorio del suelo, cuando los átomos hacen cero vibraciones alrededor de su posición más ventajosa.
- El más popular es rs estructura El subíndice "s" significa "sustitución". Lo entienden así: se cree que hay algunas coordenadas de átomos que están fijas en el espacio, luego hacen una sustitución isotópica de un solo átomo de un átomo en la molécula, y determinan la posición de este átomo cambiando las constantes rotacionales. La principal ventaja de esta tecnología es la simplicidad. Menos: solo necesita monosustitución + no todas las posiciones de los átomos se pueden establecer de esta manera + el significado físico de tal modelo no está muy claro.
- Desarrollo lógico rs las estructuras son rm -Estructuras que se obtienen del ajuste por mínimos cuadrados ponderados en masa. También necesitan moléculas sustituidas isotópicamente, pero cualquiera de ellas ya es adecuada.
Y esto está lejos de todos los tipos posibles de estructuras ...
Pero el
gran simulador El usuario de algún paquete químico cuántico estándar (como el
Programa Gaussian Evil Corporation ) cuando usa un hechizo mágico como "Opt" obtiene lo que se llama "geometría de equilibrio", o
re estructura Esta es la configuración más óptima de los núcleos, minimizando la energía electrónica del sistema. Y tales estructuras también pueden extraerse de la difracción de electrones y la espectroscopía rotacional, pero solo para moléculas muy pequeñas y simétricas, y en combinación con otros métodos de investigación. Hasta ahora no funciona.
Y entonces surge la pregunta: ¿es correcto comparar
re estructura con algunos de los experimentales, mirando solo los errores experimentales?
La respuesta aquí es simple:
no , es necesario establecer un error adicional sobre posibles diferencias sistemáticas. Y se puede dar un ejemplo muy vívido de esto: el efecto Bastiansen-Morino (ver artículos
DOI: 10.1107 / S0365110060002557 y
DOI: 10.1107 / S0365110060002545 ).
Supongamos que tenemos una molécula de tipo CX
2 (es decir, CO
2 , CS
2 , etc.). Como deberíamos saber por el curso de la química escolar, estas moléculas tienen una estructura lineal (los átomos de carbono y dos X calcogenes se encuentran en una línea recta).
Esto significa que la distancia entre los átomos X debe ser igual al doble de la longitud del enlace C - X (es decir,
re( mathrmXX)=2re( mathrmCX) )

De todos modos, si medimos las distancias entre los átomos C y X (
rg( mathrmCX) ) y XX (
rg( mathrmXX) ) por difracción de electrones gaseosos, obtenemos que
rg( mathrmXX)<2rg( mathrmCX) es decir la molécula resulta ser curva. La razón radica en el hecho de que la molécula hace el llamado
vibraciones de tijera , debido a que los átomos X están mucho más cerca uno del otro que en la ubicación más favorable (ver la figura a continuación).

¿De dónde viene el efecto Bastansen-Morino? Imagen del artículo de
DOI: 10.1039 / C6CP05849C .
Por lo tanto, si igualamos la temperatura promedio
rg -estructura al equilibrio (
re ), llegaríamos a la conclusión errónea de que las moléculas de dióxido de carbono y disulfuro de carbono son curvas.
Es por eso que al comparar diferentes tipos de parámetros geométricos, siempre debe tener mucho cuidado. Esto se aplica tanto a una comparación de datos experimentales entre ellos como a una comparación de experimento y teoría.
Moléculas Moléculas Modelo Estándar
Ahora imaginemos que deseamos sinceramente simular el resultado de algún experimento sobre la base de nuestro modelo teórico para comparar la simulación con la realidad en una batalla justa.
Y aquí también es necesario tener cuidado, porque diferentes modelos de moléculas también tienen sus límites de aplicabilidad. Examinemos esto usando el ejemplo del Modelo de Molécula Estándar.
Primero debe comprender cuál es el modelo estándar de molécula. Los físicos de BAK tienen su propio
modelo estándar , los astrónomos tienen el
suyo y los físicos tienen su propio diseño básico, del cual luego bailan. Pero a diferencia de los modelos físicos, lo que consideramos es un conjunto de aproximaciones que permiten al usuario obtener el resultado de forma relativamente automática y rápida.
Para usuarios gaussianosAhora recordamos lo que está en la base de los hechizos mágicos gaussianos "Opt" y "Freq".
El esquema general de las aproximaciones introducidas se parece a esto:
En el fondo de la calidad está nuestro modelo estándar. Revise brevemente todas las etapas de su recepción.
El modelo resultante se llama RR-HO (@BO). No tocaremos la aproximación de Born-Oppenheimer (BO), pero tendremos que hablar sobre el rotador duro y el oscilador armónico en el marco de la química estructural ...Y el principal problema con esta aproximación es que la molécula no es rígida y sus vibraciones son completamente armónicas. Por consiguiente, en realidad, necesitamos la aproximación de un rotador no rígido y un oscilador anarmónico. Y la palabra clave aquí es "anarmónico", es decir "No armónico".Hablemos de las moléculas más simples: diatómica. Hay muchos ejemplos de ellos: HCl, HBr, HI, CO, O 2 , N 2 , etc. etc.
Se distinguen de todas las moléculas por el hecho de que solo tienen una vibración: la extensión / compresión de la distancia interatómica.Y esta es la distancia entre los átomos que podemos medir en la difracción de electrones gaseosos (en una variante de la temperatura promedio,r g ) y en espectroscopía rotacional (promediada sobre, por ejemplo, el estado vibratorio del suelo, es decirr 0 )
Y ahora surge la pregunta principal del universo de la vida y en general:que sera r g y r 0 en la aproximación de un oscilador armónico, y cómo se correlaciona esto con la distancia de equilibrior e ?
Para obtener una respuesta, debe observar la superficie de energía potencial de una molécula diatómica:
- , : . , .
- , , 2 :
- ( r→0 ), - , « » ,
- ( r→+∞ )
Como resultado, si la molécula acorta su longitud de enlace en relación con la posición de equilibrio, se apoya contra la pared y, si aumenta, cae sobre un sofá blando. Y la molécula no es tonta, yacerá más en el sofá que golpeada contra la pared. Por lo tanto, la vibración promediada a lo largo de la distancia será mayor que el equilibrio (r e < r 0 , r g ), y esto es notable: tales desplazamientos son del orden de 0.01 Å, que es mayor que los errores de medición.
Por lo tanto, incluso si queremos calcular algo más como un experimento, permaneciendo dentro del marco del Modelo de Molécula Estándar (RR-HO @ BO), no obtendremos nada nuevo, por lo tanto, la geometría de equilibrio participará en la comparación.Error Conclusión # 2
 Ilustración del artículo del DOI: 10.1002 / anie.201611308 .Y la conclusión es terriblemente simple y consta de 2 partes.
Ilustración del artículo del DOI: 10.1002 / anie.201611308 .Y la conclusión es terriblemente simple y consta de 2 partes.- Con una comparación correcta, todos los valores deben tener el mismo significado.
- Si los valores son diferentes, entonces esto no debe olvidarse.
Ejemplos de errores en trabajos científicos.
"Obras hindúes"
En realidad, el lugar principal donde puedes encontrar esto es revistas de bajo nivel. Raramente contienen artículos con resultados interesantes, por lo que fueron elegidos por los "Investigadores líderes" de países del segundo y más mundos (países BRICS y sus seguidores menos exitosos). Por revistas de "bajo nivel" se entiende aquí no aquellas que publican artículos como "The Rooter: Algorithm for the Unification Típico de Puntos de Acceso y Redundancia", sino publicaciones científicas respetadas. En mi campo científico, los "medios lavados" más famosos son:(hay otros) Como puede ver, según signos formales, en la ciencia rusa se consideran publicaciones muy decentes. Pero, viene una afluencia de r ... contenido de dudosa calidad que aún se filtra mucho.Como ilustración, tomé el último número del Journal of Molecular Structure y revisé la tabla de contenido, y listo:S. Sathiya, M. Senthilkumar, C. Ramachandra Raja, crecimiento de cristales, análisis de superficie Hirshfeld, estudio DFT y estudios NLO de tercer orden de tiourea 4 dimetil aminobenzaldehído // J. Mol. Struct., V. 1180 (2019), PP. 81-88.
https://doi.org/10.1016/j.molstruc.2018.11.067
La estructura general de dicho trabajo es muy sencilla.- Alguna sustancia se "hierve" (pero con mayor frecuencia se compra estúpidamente en Sigma ). En este trabajo, la sustancia todavía se cocina.
- (), . — , ( , ) , . , - Gaussian, « ». … 1 2 , .. - .
- , /Vis, .
- En los visualizadores moleculares estándar, como GaussView (gruñido) se dibujan bellas imágenes, pero siempre en baja calidad.
- No se sacan conclusiones sustantivas: “experimentamos mucho, contamos mucho, trajimos tablas e imágenes ⇒ somos geniales, danos dulces ".
Pero de mis favoritos: artículoM. Govindarajan, M. Karabacak, FT-IR, FT-Raman y la investigación espectral UV; análisis de estimación de frecuencia calculada y cálculos de estructura electrónica en 1-nitronaftaleno // Spectrochimica Acta Parte A: Espectroscopía molecular y biomolecular, V. 85 (2012), PP. 251-260,
https://doi.org/10.1016/j.saa.2011.10.002.
En él, el espectro en la fase sólida se registró estúpidamente y luego se interpretó sobre la base de cálculos mediocres en el modelo HO en la fase gaseosa. Pero el truco es que ni siquiera podían hacer cálculos normales, lo cual insinuaron cortésmente en el comentario sobre el artículo .Sin embargo, el término "obras hindúes" (como el término " código hindú ") se refiere lejos de las únicas obras que provienen del subcontinente misterioso correspondiente.Si vas al maravilloso sitio del Cyberleninka , entonces miras el Abismo, y el Abismo te mira a ti, puedes descubrir muchas cosas interesantes. Búsqueda de "cuántico-química» (c / sin condiciones adicionales 'PCA') fue capaz de encontrar un montón de cosas que el diabloútil Dado que una gran cantidad de trabajos de "química cuántica" se dedicaron al estudio de caballos esféricos en el vacío (es decir, cálculos sin referencia a la realidad), no estaban relacionados con este texto. Pero entre ellos, estas tres obras se perdieron:Estuve especialmente satisfecho con esto último, porque la "evaluación de adecuación" fue una comparación inadecuada de estructuras en diferentes fases (gas versus cristal) y con diferentes significados ( r e vs.r α ) es de hecho la altura de la adecuación.¿Sucede en buenas revistas?
Sí, hay "errores" allí.¡No tengas miedo, todo terminó bien!, , , / . , , .
, : , .
Estamos hablando de una de las moléculas orgánicas con un enlace C - C único extremadamente largo: 1,1'-bisdiadamantano :
¿Por qué esta molécula es genial?,
(
), , C--C 1.54 Å.
, 1,1'-
re=1.630±0.005 Å, 0.08 Å ( )!
C--C - , , , , . , , () . - , . , ( ) , .
La conexión central unaria C - C es muy larga y, por lo tanto, fue muy interesante comparar cómo los métodos teóricos reproducen la realidad. Y la realidad está en los primeros artículos, aquí están:
Holy shit!— Nature, , — JACS — !!! , - .
estuvo representado solo por datos cristalográficos. Como resultado, al comparar cosas comparables difíciles entre sí, sin hacer las correcciones adecuadas para la diferencia en los parámetros, finalmente llegaron a la conclusión de que la longitud del enlace C - C central en el gas es 1.655 Å, superado por 0.02 Å. Y esto es significativamente más que el error experimental.Afortunadamente, al final, cooperaron con especialistas en estos temas, y al final recibieron la respuesta correcta ( también se puede encontrar un breve resumen popular de este trabajo en N + 1 ).¿Necesitas una comparación?
 Después de todo lo que escribí sobre la exactitud de las comparaciones, puede surgir una pregunta razonable: ¿es necesario comparar los resultados de los cálculos y los resultados de los experimentos entre ellos?Es necesario! Justo como lo necesitas!Hay una famosa declaración (de la cual no pude encontrar un autor confiable):
Después de todo lo que escribí sobre la exactitud de las comparaciones, puede surgir una pregunta razonable: ¿es necesario comparar los resultados de los cálculos y los resultados de los experimentos entre ellos?Es necesario! Justo como lo necesitas!Hay una famosa declaración (de la cual no pude encontrar un autor confiable):Nadie cree en los cálculos teóricos, excepto el que los hizo.
Todos creen resultados experimentales, excepto el que los obtuvo.
Traducido al ruso, suena como: Nadie cree los cálculos teóricos, excepto el que los hizo, pero todos creen los datos experimentales, excepto el que los recibió.Pero en ciencia es necesario que todos crean (bueno, o la mayoría), y el experimento es la única medida, ya que relaciona lo que hemos calculado con la Realidad.Hay un ensayo maravilloso (acceso abierto) en la segunda revista química más importante sobre el tema de lo que debe hacer un investigador o una persona promedio que lee artículos y / o noticias científicas: Mata R., Suhm M. // Angew. Chem Int. Ed., 56 (2017), DOI: 10.1002 / anie.201611308(por cierto, ya le di un enlace, ya que una foto, de Ricardo Mata, es de este artículo).Las conclusiones de este ensayo proporcionan recomendaciones a los teóricos y experimentadores de simulación. Los daré aquí (en traducción y una pequeña revisión) como una palabra final para esta publicación.- El teórico debe:
- no solo proporciona métodos e intentos exitosos, sino que también describe los fracasos de los métodos (especialmente si estos métodos son populares),
- describe tu metodología bien y completamente,
- donde es posible dar estimaciones (o descripciones) de errores e importantes aproximaciones y simplificaciones aceptadas.
- Los experimentadores, a su vez, deben:
- para empujar a los teóricos sus datos experimentales, que podrían usar como puntos de referencia (estándares),
- para mostrar a la comunidad científica datos experimentales incomprensibles, donde la teoría (o experimentos adicionales) ayudaría con la explicación,
- hablar en
territorio extranjero de conferencias teóricas con sus datos,
- sacar de sus datos experimentales cosas que sean lo más accesibles posible para compararlas con la teoría,
¡Todas las mejores y correctas comparaciones! Y recuerda: solo el que no hace nada no se equivoca.
PS
Como epílogo, me gustaría dar una pequeña lista de bases de datos donde puede desenterrar varios datos experimentales para moléculas.Bancos de datos estructurales
Latas cristalográficas
La forma más fácil de determinar la estructura de una molécula en un cristal es porque PCA es una rutina. Por lo tanto, si no sabe cómo se ve la molécula, vaya a los bancos de datos cristalográficos (lugares donde se recogen casi todas las estructuras de sustancias que alguna vez se amontonaron en un goniómetro e iluminadas por un haz de partículas de onda corta). Como hay muchos de estos bancos, daré solo los más famosos (se puede encontrar una lista más completa en el Wiki ).- Base de datos de estructura cristalina inorgánica ( ICSD ). No se trata completamente de moléculas, allí se pueden encontrar principalmente estructuras de diferentes sales, metales, cerámicas, etc. Esta base es apoyada por la Universidad Tecnológica de Karlsruhev, por lo tanto, el acceso a ella es de pago y no es barato. Pero si eso, su sitio .
- Cambridge Structural Database ( CSD ). , . ! . ! . , , , , . .
- Crystallography Open Database ( COD ). - . , , , , . .
- , , Protein Data Bank ( PDB ). , ( ). .
La estructura de las moléculas en un gas.
Aquí las cosas son algo peores, porque Los experimentos para estudiar la estructura de las moléculas libres son mucho más complicados, tanto para llevar a cabo (al menos se necesita un alto vacío) como para la interpretación.Por lo tanto, hay sustancialmente menos bases de datos.- La base de datos más grande para las moléculas libres es la MOlecular GAsphase DOCumentation (o MOGADOC ). Tiene su sede en la Universidad de Ulm, y es una inversión muy costosa. Pero, en todo caso, el sitio está aquí .
- Si desea conocer las estructuras de equilibrio 100% experimentales de las moléculas, esta es la Comparación de Química Computacional NIST y la Base de Datos de Referencia ( CCCBDB ). Casi todo puramente experimental.re - , . .
- — MolWiki . , , , ( ). .
?
 Tomado de xkcd.comAquí hay significativamente más bases de datos, ya que eliminar el espectro sin interpretación es una tarea mucho más simple que obtener una estructura (no es necesario construir modelos y demostrar que son correctos). Y además, los espectros son de gran valor aplicado: pueden usarse para determinar la composición de las muestras, ya sea agua de un río vecino o una señal de una nube molecular (o incluso de una atmósfera de exoplanetas, ver la ilustración de arriba).Sí, por cierto, todos los enlaces en esta sección serán a bases de datos gratuitas.
Tomado de xkcd.comAquí hay significativamente más bases de datos, ya que eliminar el espectro sin interpretación es una tarea mucho más simple que obtener una estructura (no es necesario construir modelos y demostrar que son correctos). Y además, los espectros son de gran valor aplicado: pueden usarse para determinar la composición de las muestras, ya sea agua de un río vecino o una señal de una nube molecular (o incluso de una atmósfera de exoplanetas, ver la ilustración de arriba).Sí, por cierto, todos los enlaces en esta sección serán a bases de datos gratuitas.- NIST Chemistry WebBook . , . , UV/Vis, - . , ! , , . .
- High-resolution transmission molecular absorption database ( HITRAN ). - , , , , (, ). .
- , The Cologne Database for Molecular Spectroscopy, . .
- Bueno, como último ejemplo, daré el proyecto ExoMol . Estos no son espectros experimentales completamente puros, pero este es un excelente ejemplo de la interacción de la teoría y el experimento: basado en datos experimentales de alta precisión y cálculos de muy alto nivel, los espectros de moléculas simples se predicen en condiciones diferentes (incluso extremas). El énfasis principal aquí está en los biomarcadores, por lo que cuando los astrónomos ven los espectros de los exoplanetas, pueden identificar fácilmente las moléculas que ya conocemos en ellos. Sitio .
PPS
Si hay errores / algo sigue siendo incomprensible, escriba en los comentarios; lo corregiré / intentaré explicar mejor.