Lorsque le morphologue britannique George Jackson Mywart [St. George Jackson Mivart] a publié en 1865 l'un des premiers arbres évolutionnaires, il manquait de matériel de support. Il a construit un arbre - une
carte ramifiée de diverses espèces de primates - en utilisant une analyse détaillée des épines animales. Le deuxième arbre, basé sur une comparaison de membres d'animaux, a
montré d'autres liens familiaux entre primates, mettant en évidence le problème de la biologie évolutive qui existe à ce jour.
Près de 150 ans plus tard, les scientifiques ont acquis des montagnes de données pour construire les soi-disant
arbres phylogénétiques , une version moderne de la structure créée par Mivart. Les progrès de la technologie de décodage de l'ADN et de la
bioinformatique vous permettent de comparer les séquences de centaines de gènes, et parfois des génomes entiers, d'espèces différentes, et de créer l'arbre de vie avec plus de détails que jamais.
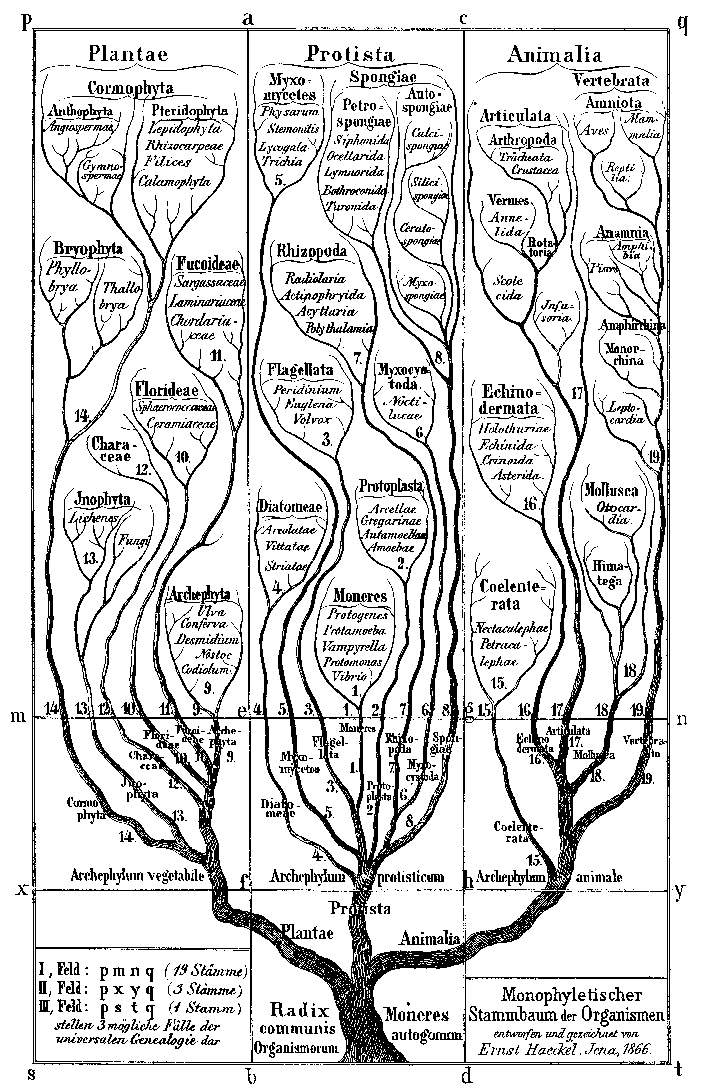 L'arbre de vie historique de 1866 décrit les royaumes des plantes, des animaux et des cellules unicellulaires
L'arbre de vie historique de 1866 décrit les royaumes des plantes, des animaux et des cellules unicellulairesMais bien que l'abondance de données ait aidé à résoudre certains des conflits qui ont surgi sur différentes parties de l'arbre évolutif, cela a également apporté de nouvelles difficultés. La version d'aujourd'hui de l'arbre de vie ressemble plus à une page Wikipedia controversée qu'à un livre publié - il y a des débats en cours sur certaines branches. Tout comme la colonne vertébrale et les membres ont conduit à l'émergence de cartes contradictoires de l'évolution des primates, les scientifiques savent maintenant que différents gènes dans le même corps peuvent raconter des histoires différentes.
Selon une nouvelle étude, basée en partie sur l'étude de la levure, l'image controversée dessinée par les gènes individuels est encore plus contradictoire que prévu. «On prétend que chacun des 1070 gènes est impliqué dans une sorte de conflit», explique Michael
Donoghue , biologiste évolutionniste à Yale qui n'est pas associé à l'étude. «Nous essayons de comprendre les relations phylogénétiques de 1,8 million d'espèces et nous ne pouvons pas trier nous-mêmes vingt types de levures», dit-il.
Pour résoudre le paradoxe, les chercheurs ont développé un algorithme basé sur la théorie de l'information pour mesurer le niveau de confiance dans l'exactitude de certaines parties de l'arbre. Ils espèrent que la nouvelle approche aidera à clarifier les périodes d'évolution qui possèdent à la fois les données les plus intéressantes et utiles et les plus conflictuelles - par exemple, l'
explosion cambrienne - la diversification rapide de la vie animale qui s'est produite il y a 540 millions d'années.
"Historiquement, les épisodes les plus intéressants sont liés à des domaines qui ont attiré l'attention et ont suscité la controverse", comme l'origine des animaux, des vertébrés et des plantes à fleurs, explique
Antonis Rokas , biologiste à l'Université Vanderbilt, qui a dirigé la nouvelle étude.
Sur la base des résultats du nouvel algorithme, les scientifiques ne peuvent sélectionner que les gènes les plus informatifs pour la construction d'arbres phylogénétiques. Une telle approche peut rendre le processus à la fois plus précis et plus efficace. «Je pense que cela aidera à accélérer la reconstruction de l'arbre de vie», a déclaré Khidir Hilu, biologiste au Virginia Institute of Technology.
Briques de vie
La base des arbres phylogénétiques est créée par le regroupement des espèces en fonction de leur degré de parenté. Si nous comparons l'ADN des humains, des chimpanzés et des poissons, il devient clair que les humains et les chimpanzés sont plus proches les uns des autres que des poissons.
Il était une fois, les chercheurs utilisaient un ou plusieurs gènes pour comparer des organismes. Mais au cours de la dernière décennie, il y a eu une explosion de données phylogénétiques, remplissant très rapidement les bases nécessaires à la création de ces arbres. L'analyse a rempli plusieurs des taches blanches dispersées à travers l'arbre, mais de sérieux désaccords existent toujours.
Par exemple, on ne sait pas encore qui est le plus proche en nature des escargots - les mollusques bivalves ou
les mollusques à pattes de pelle , dit Rokas. On ne sait pas exactement comment certaines des premières branches d'animaux d'un arbre, comme les méduses et les éponges, sont interconnectées. Les scientifiques peuvent montrer des exemples d'arbres conflictuels apparaissant dans les mêmes revues scientifiques avec une différence de semaines, ou
même dans le même numéro .
"D'où la question: pourquoi est-il si difficile pour nous d'être d'accord?" - dit Rokas.

Rokas et son étudiant diplômé Leonidas Salichos ont étudié cette question en
évaluant les gènes individuellement , en utilisant les gènes les plus utiles - portant le plus d'informations liées à l'histoire de l'évolution - pour construire leur version de l'arbre.
Ils ont commencé avec 23 espèces de levures et sélectionné 1 070 gènes. Pour commencer, ils ont créé un arbre phylogénétique de manière standard, la concaténation. Pour ce faire, toutes les séquences d'espèces individuelles sont rassemblées en un mégagène, puis les séquences d'espèces individuelles sont comparées à cette longue séquence, sur la base de laquelle un arbre est créé qui explique le mieux les différences.
L'arbre résultant est précis en termes d'analyse statistique standard. Mais comme des méthodes similaires conduisent à des arbres grouillant de désaccords, Rokas et Salichos ont décidé de se plonger dans le sujet. Ils ont construit des ensembles d'arbres phylogénétiques pour des gènes de levure individuels et appliqué un algorithme développé en utilisant la théorie de l'information pour rechercher les zones de plus grande correspondance entre différents arbres. Le résultat,
publié dans la revue Nature en mai , était inattendu. Chaque gène étudié semble raconter une histoire évolutive légèrement différente.
«Presque tous les arbres construits pour des gènes individuels se sont heurtés à un arbre basé sur la concaténation de données», explique Hilu. "C'est choquant."
Ils ont conclu que si plusieurs gènes prennent en charge une architecture particulière, elle doit être précise. Mais si différents ensembles de gènes prennent également en charge deux architectures différentes, la probabilité de leur correspondance exacte avec la réalité est réduite. Rokas et Salichos ont utilisé une méthode appelée
bootstrap statistique pour sélectionner les gènes les plus informatifs.
En fait, "si vous prenez uniquement des gènes avec un support actif, vous obtiendrez le bon arbre", explique Donogue.
L'arbre révisé a coïncidé avec un arbre construit sur une source alternative d'informations évolutives - des changements à grande échelle dans les segments d'ADN transmis de génération en génération - qui ont justifié leurs recherches.
Les découvertes ne se sont pas limitées à la levure. En appliquant la même analyse à des formes de vie plus grandes et plus complexes, y compris les données génétiques des vertébrés et des animaux, ils ont trouvé de graves conflits entre les gènes individuels.
Certains chercheurs doivent s'habituer à l'idée d'exclure sélectivement les données de l'analyse. «Pendant de nombreuses années, le principal problème pour les personnes qui tentent de comprendre les relations entre les organismes a été le problème de la collecte de suffisamment de données», explique
Jeffrey Townsend , biologiste évolutionniste à Yale qui n'est pas lié à la recherche. «La communauté a toujours été informée de la nécessité d'un ensemble de données, il n'est donc pas surprenant qu'elle ait abordé la tâche de cette façon.»
Bien que les biologistes évolutionnaires aient lutté avec ces problèmes pendant des années, la nouvelle étude est devenue la plus grande tentative à ce jour pour étudier le niveau de conflit des gènes individuels. «Les gens auront deux réactions: il y a plus de conflits que je ne le pensais, et nous devons apprendre à mieux les analyser», explique Donague, qui souhaite utiliser la nouvelle méthode dans son travail. Cependant, il souligne également des difficultés à confirmer l'exactitude de la nouvelle approche. Bien que l'arbre révisé coïncide avec ce qui est construit sur des informations génétiques alternatives, ce dernier peut révéler ses propres incohérences. «Je ne suis pas sûr que nous sachions quelle est réellement la relation», dit-il. "Et si nous ne sommes pas sûrs du véritable état des choses, nous ne savons pas si nous avons le bon arbre."
Changer l'image
Les chercheurs doivent appliquer la nouvelle technique plus largement pour voir comment elle peut changer le concept d'évolution. Cependant, Rokas et Salichos ont déjà montré qu'il est plus difficile de reconstruire de courtes branches de l'arbre, ou des parties «buissonnantes» de celui-ci, représentant des périodes de spéciation rapide - en particulier celles situées plus près de la base de l'arbre et profondément dans l'histoire de l'évolution.
"La recherche théorique a prédit ce comportement, mais notre étude pour la première fois démontre une confirmation en utilisant des données expérimentales", a déclaré Rokas.
Rokas soutient que les nouvelles découvertes changeront la façon dont les chercheurs interprètent les parties d'un arbre mal définies. «Les biologistes évolutionnistes supposent généralement que si l'arbre n'a pas les détails nécessaires, alors c'est faux. Et donc, si nous collectons plus de données et composons de meilleurs algorithmes, alors nous arriverons à la bonne arborescence », dit-il. Mais la présence de parties conflictuelles de l'arbre qui persistent, malgré les flux de données et l'application d'un nouveau type d'analyse, peut indiquer la présence de parties touffues. «Je pense que dans certains cas, l'algorithme sera en mesure de résoudre ce conflit, et dans d'autres, il est possible de marquer les zones de conflit que nous ne pourrons probablement pas résoudre.
L'étude de ces parties touffues de l'arbre peut donner un nouveau regard sur les étapes particulièrement intéressantes de l'évolution, par exemple, l'explosion cambrienne, lorsque la vie est passée de la prédominance d'organismes simples à un ensemble varié d'espèces animales.
D'autres chercheurs conviennent que les découvertes peuvent influencer la façon dont les spécialistes traitent les idées contradictoires sur l'évolution. "Je pense que c'est un signe avant-coureur d'un changement de paradigme", a déclaré Townsend. «Si nous utilisons des méthodes appropriées, nous aurons l'occasion d'en apprendre davantage sur des problèmes qui nous tourmentent depuis longtemps.»
Townsend, qui a développé sa propre méthode de sélection des gènes les plus informatifs en fonction de
leur taux d'évolution , note que tous les membres de la communauté scientifique ne sont pas d'accord sur la nécessité de nouvelles approches. «J'espère que ce travail contribuera à mettre ce problème au premier plan», a-t-il déclaré.
Choisir la bonne quantité de gènes pour construire des prototypes d'arbres phylogénétiques n'est pas la seule question qui afflige les biologistes évolutionnaires. Ils doivent également s'entendre sur le nombre d'espèces à inclure dans le traitement - plus il y a d'espèces dans l'arbre, plus l'analyse est difficile. Les résultats peuvent également varier en raison de différences dans la qualité des données collectées pour différentes espèces. «Si nous devons obtenir une véritable histoire évolutive de la façon dont tout est connecté les uns aux autres, alors quoi de mieux pour cela - pour collecter plus de gènes ou plus d'espèces? - dit Donogue. "Je pense que les deux."
De nouvelles approches qui permettent aux chercheurs d'obtenir des résultats précis en utilisant moins de gènes peuvent étendre l'arbre évolutif. La possibilité de sélectionner uniquement les gènes les plus informatifs peut rendre le processus plus efficace et permettre aux scientifiques de créer des arbres précis en utilisant moins de données et de ressources. «Si nous pouvions sélectionner plusieurs gènes et obtenir le même bon arbre que pour tout le génome», explique Khilu, «nous pourrions construire un arbre de vie beaucoup plus détaillé - au niveau des genres, voire au niveau des espèces - au lieu de nous contenter de le squelette des branches les plus importantes. "