L'analyse de l'appartenance à la population humaine par l'ADN, selon notre expérience, soulève trois grandes questions parmi le public: les gènes et les groupes ethniques peuvent-ils être liés entre eux, comment l'analyse de l'origine d'un point de vue technique et si les tests génétiques peuvent «identifier les Juifs». Pour une raison quelconque, c'est précisément la question de l'identité juive avec l'ADN qui inquiète à la fois ceux qui ont des preuves indéniables d'appartenance au peuple choisi par Dieu et ceux qui ne mangent pas de matzo et ne lisent pas la Torah.
Dans le nouveau matériel Genotek sur Geektimes, nous allons essayer de répondre à tout dans l'ordre. Et oui, nous définissons également les Juifs.

Races aka groupes de population en biologie, médecine et génétique
L'humanité a la mauvaise habitude de justifier la violence par la supériorité «innée» d'une race sur une autre - c'est pourquoi les biologistes modernes abordent la question des différences génétiques entre les populations avec une plus grande prudence. L'existence (inexistante) de frontières biologiques entre les groupes raciaux et ethniques a été discutée avec véhémence tout au long du XXe siècle, mais aucun consensus final n'a encore été atteint sur cette question (
1 ).
On espérait que le séquençage du génome humain rassemblerait tout le monde. Le génome, lu «de» et «à», montrera que les frontières entre les groupes sont de nature sociale et que les gènes sont les mêmes pour tout le monde. Cela s'est avéré différemment: une étude minutieuse du code des nucléotides humains a ravivé et un intérêt accru pour les différences biologiques entre les populations raciales et ethniques. Les mêmes gènes, en général, ont trouvé des variantes alléliques légèrement différentes associées au risque de maladies (
2 ), au métabolisme des médicaments (
3 ), à la réponse du corps aux conditions environnementales (
4 ), et ces variantes ont été trouvées dans différentes populations avec des fréquences différentes.
La recherche de gènes "indiens" ou "africains" inexistants a été arrêtée, mais la recherche dans le domaine de la génétique médicale et des populations établit encore des parallèles entre les caractéristiques biologiques et l'origine ethnique des participants. L'utilisation des termes «race» et «ethnicité» dans ces œuvres est activement débattue (et souvent condamnée). On a tenté d'introduire des règles obligeant les chercheurs à justifier la nécessité d'utiliser des catégories «glissantes» et à clarifier ce que l'on entend exactement par des termes spécifiques. En février de l'année dernière, Science, l'une des revues scientifiques les plus respectées, a publié un article ambigu (
5 ), proposant d'abandonner complètement l'utilisation du terme «race» dans la recherche génétique, en le remplaçant par une «ascendance» - «origine» plus correcte et plus neutre. .
Mais même dans des conditions d'incertitude avec les termes, l'humanité est toujours divisée en groupes de population: en particulier, pour la bonne conduite des essais cliniques de médicaments et l'évaluation du risque de maladies. Par exemple, trois variantes alléliques du gène NOD2 - R702W, G908R et 1007fs - sont associées à un risque accru de maladie de Crohn chez les Américains européens (
6 ,
7 ), cependant, aucune de ces variantes n'est associée à la maladie de Crohn en japonais (
8 ). Les allèles du gène CCR5 sont connus pour affecter le taux de développement de l'immunodéficience chez les patients infectés par le VIH (
9 ): une option a été trouvée parmi eux qui ralentit la progression de la maladie chez les Américains d'origine européenne, mais accélère son développement chez les Afro-Américains (
10 ). Les Asiatiques ont trouvé une corrélation entre les polymorphismes du gène de la protéine p53, qui régule la réponse au stress et supprime le développement des tumeurs, et les températures hivernales moyennes dans les habitats des populations - adaptation génétique au gel (
11 ). Et si, dans le passé, seules les informations fournies par les participants eux-mêmes étaient utilisées pour décomposer l'échantillon en groupes ethniques, dans l'ère post-génomique, elles sont de plus en plus complétées et affinées par une évaluation génétique de l'origine du sujet.
Variation génétique entre les populations
Dans la vie de tous les jours, nous divisons les gens en groupes selon l'apparence ou le langage de communication. La plupart des Danois se ressemblent plus que chacun d'eux ne ressemble à un Italien (
voici une visualisation cool avec des portraits moyens de différentes nationalités). Les Danois et les Italiens sont beaucoup plus proches les uns des autres que chacun d'eux - des habitants de l'Afrique subsaharienne: les phénotypes humains sont regroupés selon le modèle géographique. La distribution des génotypes a une structure similaire: les membres d'un groupe local, en règle générale, ont des liens familiaux plus étroits que les résidents des régions éloignées, et les populations vivant dans une région sont plus proches que celles dont les habitats sont séparés par des barrières géographiques (par exemple, les chaînes de montagnes ou l'eau tableau).
De plus, la diversité génétique de la population humaine est inférieure à celle de nombreuses espèces biologiques. Cela s'explique par le fait que l'humanité est une espèce jeune: les groupes individuels ont eu relativement peu de temps pour accumuler les différences. Deux personnes choisies au hasard diffèrent l'une de l'autre par ~ 1000 nucléotides, tandis que les deux chimpanzés ne coïncident pas une fois en ~ 500 "lettres". Et pourtant, au total, il y a environ 3 millions de «points de différence» potentiels dans le génome humain. La plupart de ces écarts, appelés polymorphismes mononucléotidiques (SNP), sont neutres ou presque neutres, mais certains d'entre eux sont responsables de différences phénotypiques entre les personnes.
La répartition des polymorphismes neutres (puisqu'ils n'ont pas de signification biologique, ils ne sont pas soumis à une sélection évolutive directionnelle, ils sont portés par le vent des migrations) dans la population mondiale reflète l'histoire démographique de notre espèce. Les preuves génétiques et archéologiques indiquent qu'au cours des 100 000 dernières années, la taille de la population humaine a considérablement augmenté. Les gens se sont installés en dehors de l'Afrique, colonisant le reste du monde. Le processus de réinstallation a affecté la répartition géographique des allèles de deux manières: premièrement, «l'effet fondateur» a affecté - dans la population d'immigrants, en règle générale, seule une partie des variantes génétiques de l'ensemble de leur diversité dans la population ancestrale était représentée; deuxièmement, le soi-disant "croisement assortiment" a eu lieu, c'est-à-dire les couples se sont formés principalement au sein de leur groupe, ce qui a limité la distribution des polymorphismes de novo existants et émergents parmi les individus habitant différentes zones géographiques. Ces processus ont conduit à l'accumulation progressive des différences génétiques.
Dans le contexte des groupes de population, les marqueurs génomiques ont commencé à être étudiés dans les années 70-80, dans les années 90, ils ont commencé à être utilisés pour identifier la population d'une personne en particulier. Les chercheurs ont démontré à plusieurs reprises que les polymorphismes génétiques peuvent isoler avec succès des groupes de population et déterminer l'appartenance à un groupe d'un individu. Ensuite, il a été démontré que les personnes vivant sur le même continent sont génétiquement plus proches les unes des autres que les personnes de différents continents. Initialement, dans ces études, les informations sur le lieu de naissance, la race, le groupe ethnique étaient connues dès le début et étaient utilisées conjointement avec des données génétiques; si les sujets étaient répartis à l'aveugle parmi les grappes uniquement sur la base de traits génétiques, la correspondance entre l'origine géographique, l'ethnicité et la structure de la population était moins prononcée. Comme l'ont montré d'autres études, le succès dépendait des marqueurs génétiques utilisés et de leur nombre (plus c'est mieux), du bon choix des populations de référence et d'autres facteurs (
12 ).
En 2004, aux États-Unis, la définition génétique de la population a été utilisée non seulement dans la recherche biomédicale, mais aussi dans les enquêtes criminelles:
cet article de Nature contient une histoire passionnante sur la façon dont les policiers, désespérés de trouver un criminel, ont ordonné un test ADN à une entreprise commerciale, décidé couleur de peau du suspect et a ouvert le dossier. Les suggestions pour l'analyse de l'origine génétique ont réussi à frapper la vague d'intérêt général chez les personnes dans leur propre passé. "Roots mania", ainsi appelé ce passe-temps dans un article de Time, consacré à "la dernière obsession de l'Amérique" - la recherche généalogique.
Les méthodes génomiques sont activement utilisées par des spécialistes qui étudient l'origine et l'évolution des peuples. Par exemple, en 2013, une équipe internationale de chercheurs a utilisé l'analyse génétique pour réfuter l'hypothèse de l'origine des Juifs ashkénazes des Khazars (
13 ). L'ensemble de données génomiques utilisé par les auteurs est du domaine public: il contient plus de 100 populations mondiales. Nous proposons de simuler une petite étude avec nous: pour déterminer la place des clients Genotek dans cet échantillon, et en même temps pour comprendre les détails techniques de la détermination de la population.
Objectif de la recherche
Identifiez les clients Genotek parmi les populations de référence. Découvrez s'il y a des représentants des Juifs ashkénazes dans notre échantillon. Démontrer les principes et les méthodes d'analyse de la population d'un individu.
Objectifs de recherche
Traiter les données de génotypage de 722 sujets en utilisant le programme ADMIXTURE en utilisant l'ensemble de données de Behar et al., 2013 comme échantillon d'apprentissage.
Matériel et méthodes
Les travaux originaux de Behar et al., 2013 ont utilisé des données de 1774 personnes: parmi eux, des représentants de 88 populations non juives (d'Arabie, d'Asie centrale, d'Asie de l'Est, d'Europe, du Moyen-Orient, d'Afrique du Nord, de Sibérie, d'Asie du Sud et Afrique saharienne) et 18 populations juives. Les auteurs avaient besoin d'un vaste ensemble de données pour déterminer avec précision la place des ashkénazes dans le contexte des populations mondiales: la tâche était de présenter les trois régions géographiques d'où ce groupe pourrait hypothétiquement provenir: l'Europe, le Moyen-Orient et le Khazar Khaganate. Les auteurs ont souligné la différence entre l'approche d'échantillonnage, représentant les populations modernes européennes, moyen-orientales et juives - descendants directs de populations ancestrales, et les échantillons correspondant au Khazar Kaganate, qui a cessé d'exister il y a environ 1000 ans. Le hic, c'est qu'aucune des populations existantes n'est l'héritière directe du Khaganate. En tant que représentants modernes possibles des Khazars, les auteurs ont choisi les habitants du Caucase du Sud (Abkhazes, Arméniens, Azerbaïdjanais, Géorgiens), du Caucase du Nord (Adygues, Balkars, Tchétchènes, Kabardins, Ossètes et plusieurs autres nationalités), Tchouvachie et Tatars.
Nous avons ajouté des échantillons de 722 personnes de différentes régions de Russie à l'ensemble de données.
Pour l'analyse statistique, nous avons utilisé le programme ADMIXTURE, qui nous permet d'estimer l'origine la plus probable d'un individu à partir de données sur les génotypes. En plus de cela, les auteurs de l'article en discussion ont utilisé d'autres méthodes statistiques qui ont donné une réponse similaire à la question posée. Nous nous concentrerons sur ADMIXTURE, car c'est cet algorithme qui permet d'estimer le pourcentage de contribution des populations ancestrales aux génomes étudiés.
ADMIXTURE utilise les méthodes de Monte Carlo dans les chaînes de Markov (chaîne de Markov Monte Carlo, MCMC). Voici un
lien vers un article des auteurs de l'algorithme pour ceux qui veulent comprendre plus en détail le côté mathématique du processus.
Voyons comment ADMIXTURE fonctionne sur l'exemple d'échantillons et de populations de notre set
Au total, nous avons 2 496 échantillons / individus, chacun appartenant à l'une des 106 populations modernes. Nous suggérons que les populations modernes descendent très probablement d'un nombre relativement restreint de populations ancestrales. Les «populations ancestrales» dans cette analyse sont d'anciens clusters génomiques, unis par le principe de similitude génétique. ADMIXTURE permet à la fois de proposer arbitrairement des hypothèses sur le nombre de ces grappes dans l'échantillon, et de sélectionner le nombre optimal qui décrit le mieux la distribution réelle des données génomiques.
Ayant reçu des informations sur les génotypes et le nombre estimé de populations «ancestrales» (K), ADMIXTURE construit un modèle qui estime la contribution de chacune des populations «ancestrales» à chaque échantillon. Lors de l'interprétation des données, la composition quantitative du génome (pourcentage de grappes) et qualitative sont importantes - leur présence ou leur absence dans des génomes spécifiques. Sur la base de ces données, on peut faire des hypothèses sur les processus évolutifs dans une population, en particulier, sur la présence ou l'absence de «racines» communes dans les groupes de population. Cependant, les conclusions seront légitimes si le modèle que nous avons construit est bon: la valeur optimale de K. est sélectionnée.
Nous sélectionnons la valeur optimale de K
Comment déterminer combien de populations «ancestrales» correspondent le mieux à la vérité pour un échantillon donné? Empiriquement!
ADMIXTURE est un programme intelligent: construire un modèle de la structure génétique des populations à partir de données sur des génotypes individuels (évaluer la contribution de chacun des anciens clusters génomiques à chacun des échantillons de génomes) pour un nombre K donné, elle n'oublie pas de faire une comparaison avec la réalité au final. Vérifiez dans quelle mesure l'entrée est décrite par le modèle construit. Une mesure de comparaison est l '«erreur» - une valeur qui décrit l'inadéquation entre le modèle et les données réelles. Plus l'erreur est grande, plus l'hypothèse du nombre de populations ancestrales est vraie.
Comment choisir la valeur optimale de K? Nous démarrons l'algorithme ADMIXTURE sur cet échantillon, en substituant différentes valeurs de K, et nous obtenons pour chaque K sa propre valeur d'erreur. Nous traçons la dépendance de l'ampleur de l'erreur sur K. Voici le graphique obtenu par les auteurs de l'article:
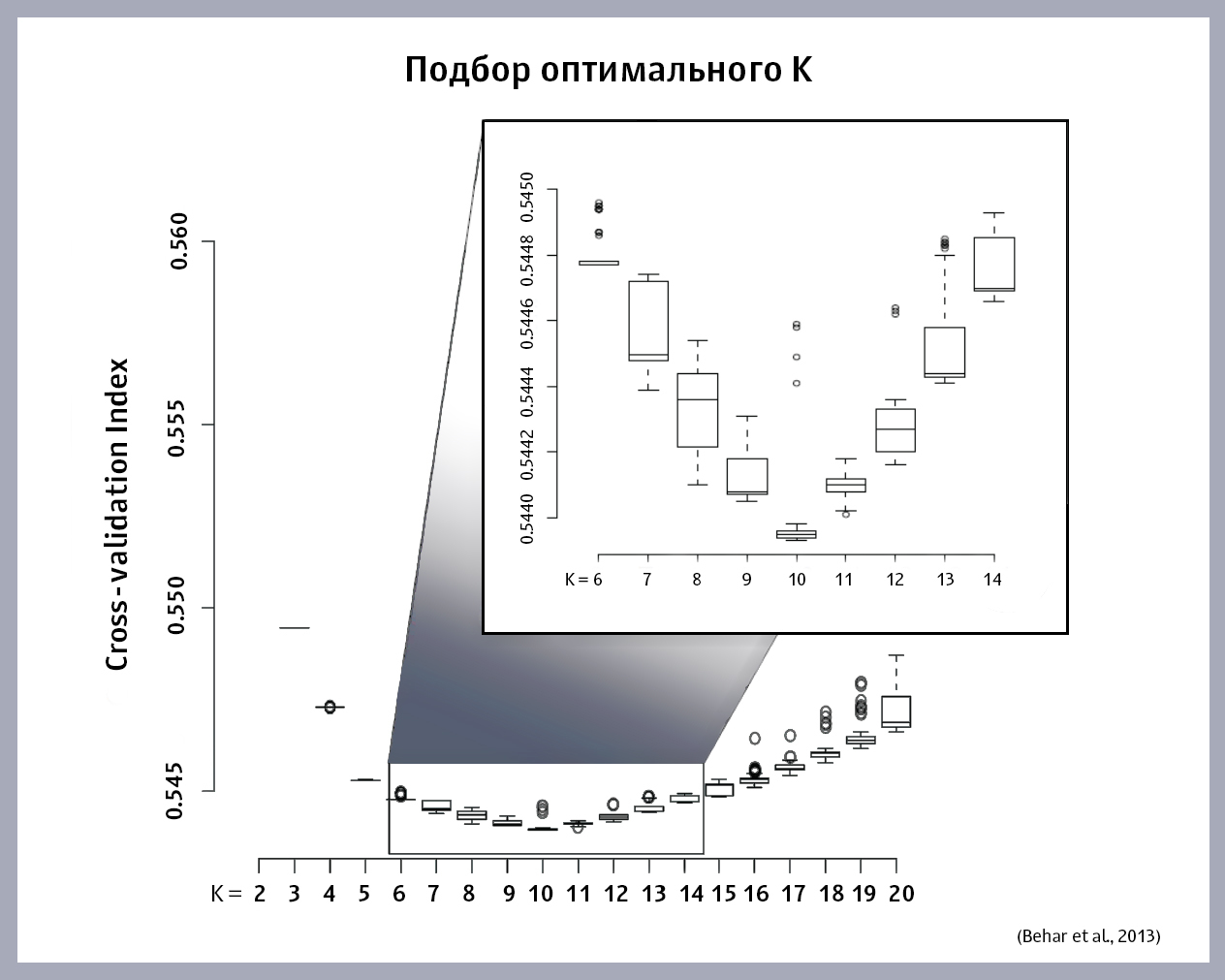
La valeur optimale de K est au point minimum de la fonction. Si le minimum n'est pas trouvé sur le graphique (la fonction augmente ou diminue constamment), vous devrez construire des modèles en choisissant de nouveaux K jusqu'à ce que vous trouviez le bon.
Même avec un K sélectionné de manière optimale, la fiabilité des résultats d'analyse dépend de l'exactitude de l'échantillon:
1. Les individus ne doivent pas être liés les uns aux autres.
2. Les polymorphismes mononucléotidiques (SNP) utilisés pour le génotypage doivent être répartis uniformément sur le génome avec une densité suffisamment élevée.
3. Les allèles SNP doivent être en liaison d'équilibre, c'est-à-dire que la probabilité de la présence d'un allèle donné chez un individu particulier ne devrait dépendre que de la fréquence de cet allèle dans la population et non des autres allèles du génome.
Comme le montre le graphique, le K optimal pour cet échantillon était de 10 populations «ancestrales».
Résultats
Les résultats de l'analyse sont visualisés par ADMIXTURE comme ceci (sur la figure, seule une partie des données est visible):
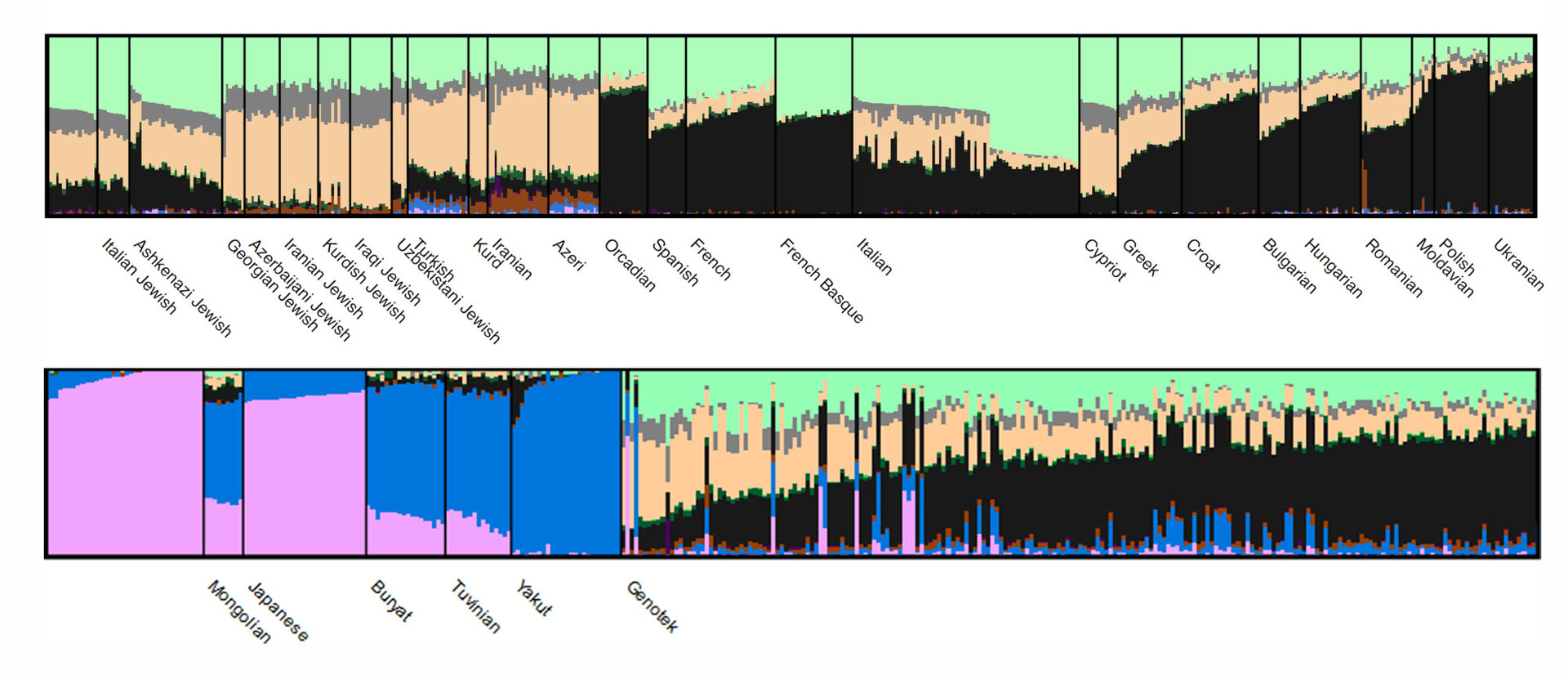
Chaque cluster a sa propre couleur, et les populations diffèrent (ou ne diffèrent pas) dans les parts des clusters dans le génome.
Ici se trouve la version interactive de l'image pour une étude détaillée: déplacez la souris et faites défiler pour voir toutes les populations ou pour examiner certains des groupes plus en détail.
En général, au sein de la «population» de Genotek, le ratio de grappes devrait correspondre au schéma caractéristique des populations d'origine est-européenne. L'intéressant commence au niveau des échantillons individuels:
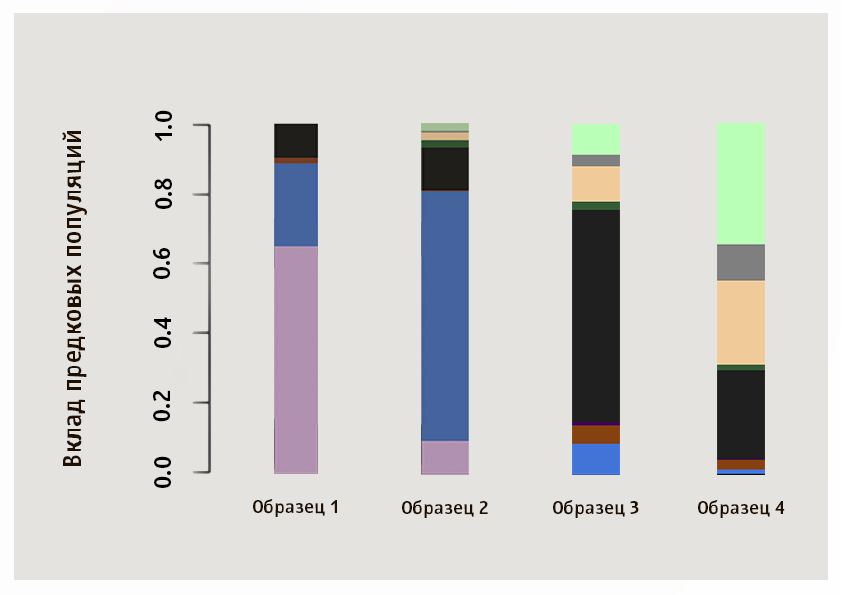
Bien que la population la plus proche de l'échantillon donné soit déterminée par des valeurs numériques, de nombreuses informations peuvent également être obtenues par comparaison visuelle des tendances. Nous vous suggérons de déterminer indépendamment les populations les plus proches pour les échantillons de quatre clients Genotek à partir de l'image.
La réponseDans cette image, les échantillons 1 et 2 sont d'origine asiatique: la prédominance de la grappe rose est typique pour les Japonais et les Khan dans notre échantillon, bleu pour les Yakuts, le troisième échantillon montre le rapport des composants typiques des Russes, Biélorusses, Ukrainiens et Polonais, et le quatrième est typique Juif ashkénaze.
Sur 722 échantillons, nous avons trouvé 9 Juifs ashkénazes.
Conclusion
L'appartenance à la population est loin d'être le seul facteur déterminant l'auto-identification ethnique d'une personne. Cependant, il est encore possible de révéler une corrélation entre les groupes ethniques et la structure du génome de leurs représentants. Une telle analyse est utilisée à la fois à des fins scientifiques et médicales, et pour l'étude de leurs propres racines par tous les arrivants. Dans le même temps, il est important de comprendre que les modèles sont constamment améliorés, et les résultats obtenus pour une plus grande précision doivent être considérés en conjonction avec d'autres données, par exemple, l'arbre généalogique de la famille.
Aucune preuve n'a été trouvée de l'origine Khazar d'Ashkenazi par les auteurs de l'article original. Les tests génétiques, bien sûr, «savent» comment identifier les Juifs - cependant, il ne faut pas oublier que le «judaïsme» est, avant tout, un état d'esprit.
Dans un avenir proche, le test ADN de généalogie mis à jour avec des résultats étendus sera lancé à Genotek: nous allons porter le nombre de populations à des centaines, ajouter des populations juives. Nous mettrons à jour les informations de votre compte personnel pour tous ceux qui nous ont déjà transmis leur matériel génétique. Si vous n'êtes toujours pas génotypé, nous vous invitons à vous
inscrire .
Les références
- Foster M., Sharp R. (2002). Race, ethnicité et génomique: classifications sociales en tant que proxys de l'hétérogénéité biologique. Genome Res.
- Collins FS, McKusick VA (2001). Implications du projet du génome humain pour la science médicale. JAMA.
- Nebert DW, Menon AG (2001) Pharmacogenomics, ethnicity, and susceptibility genes. Pharmacogénomique J.
- Olden K., Guthrie J. (2001). Génomique: implications pour la toxicologie. Mutat. Res.
- Yudell M., Roberts D., DeSalle R., Tishkoff S. (2016). Sortir la race de la génétique humaine. Science.
- Ogura, Y. et al. (2001). Une mutation de décalage de cadre dans NOD2 associée à une sensibilité à la maladie de Crohn. La nature.
- Hugot, JP et al. (2001). Association de variantes répétées riches en leucine NOD2 avec une sensibilité à la maladie de Crohn. La nature.
- Inoue, N. (2002). Manque de variantes communes de NOD2 chez les patients japonais atteints de la maladie de Crohn. Gastroentérologie.
- Martin, MP et al. (1998). Accélération génétique de la progression du SIDA par une variante promotrice de CCR5. Science.
- Gonzalez, E. et al. (1999). Effets modifiant la maladie du VIH-1 spécifiques à la race associés aux haplotypes CCR5. Proc. Natl Acad. Sci. USA
- Shi, Hong et al. (2009). La température hivernale et les UV sont étroitement liés aux changements génétiques dans la voie du suppresseur de tumeur p53 en Asie de l'Est. Journal américain de génétique humaine.
- Bamshad M., Wooding S., Salisbury B. et al. (2004). Déconstruire la relation entre la génétique et la race. Nat Rev Genet.
- Behar DM et al. (2013). Aucune preuve provenant de données à l'échelle du génome d'une origine khazar pour les Juifs ashkénazes. Biologie humaine