 Vous pouvez dire autant que vous le souhaitez qu'il y a un problème avec la médecine en Russie, mais notre protection des consommateurs est l'une des meilleures au monde.
Vous pouvez dire autant que vous le souhaitez qu'il y a un problème avec la médecine en Russie, mais notre protection des consommateurs est l'une des meilleures au monde. Comparé à nos normes européennes pour les dispositifs médicaux, ce n'est qu'un filkin.
Ces dernières années, nous devons prouver tout effet. Autrement dit, si vous rencontrez une pommade de rides, mais il ne supprime pas les rides, vous pouvez vous plaindre à Rospotrebnadzor. Et quelque part loin dans l'industrie, vous pouvez entendre le son d'un pot de gelée de pétrole en train d'être ouvert. Si vous vous sentez malade après avoir pris un baume de guérison, vous pouvez également vous plaindre. Et encore - un ppl, ils viendront à quelqu'un avec un chèque.
Toute cette histoire de durcissement des exigences nous a sérieusement affectés il y a quelques années lorsque nous avons enregistré Blefarogel. Mais permettez-moi de vous dire en détail combien de gelée de pétrole, de larmes et de morve nous avons dépensé pour ce processus.
4 cercles d'enfer
La base de l'enregistrement des dispositifs médicaux est le
décret n ° 1416 .
Il décrit les règles et la procédure d'enregistrement. Afin de subir cette procédure assez compliquée, les tests suivants doivent être effectués:
- Toxicologique (prouvant que le produit est sûr en termes de matériaux et substances utilisés dans la formule).
- Technique (prouvant que le produit, dans ses propriétés et caractéristiques techniques, répond aux exigences du TU et de la documentation opérationnelle du fabricant, ainsi qu'au fait que le processus de production sera uniforme et qu'il ne sera pas nécessaire de vérifier chaque élément individuel du lot)
- Clinique (prouvant l'efficacité et la sécurité de la résolution d'un problème médical).
- Et un examen de la qualité, de l'efficacité et de la sécurité, où la commission recueille toutes les données sur le produit en un seul tas et décide de la possibilité de son enregistrement et de sa mise sur le marché.
Un point important:
selon la loi, un dispositif médical ne peut pas guérir. Autrement dit, s'il a un effet thérapeutique, ce médicament est d'une catégorie complètement différente. Le dispositif médical peut être quelque chose d'auxiliaire dans le processus. Par exemple, si un tonomètre est enregistré, il doit être prouvé qu'il mesure la pression, que le manchon aux points de contact avec la peau ne provoquera pas d'allergies ou de dermatites diverses, que la pression sera mesurée avec une tolérance donnée tout au long de la vie de l'appareil. Dans le cas d'un canard, il est nécessaire de prouver son hypoallergénicité et sa sécurité pour le patient. Dans le cas de nos gels, l'essentiel est de passer par Roszdravnadzor, y compris la toxicologie.
La toxicologie était relativement simple. Aujourd'hui, nous devons commencer par l'analyse des composants et leurs interactions. Ce qui est important, cela concerne non seulement le produit lui-même, mais aussi l'emballage. Auparavant, ils ne vérifiaient que le gel, et maintenant, selon de nouvelles normes, le gel et l'emballage (bouteille avec un couvercle) de tous les emballages et couleurs utilisés, ainsi que leur interaction. Ceci est fait pour que la substance du flacon ne modifie pas les propriétés du gel, migrant dans sa composition.
Vous devez d'abord effectuer une revue de la littérature, c'est-à-dire trouver des informations sous forme d'articles et de critiques sur les analogues enregistrés qui prouvent leur efficacité et leur sécurité, trouver des pratiques sur les substances actives et prouver qu'elles sont sûres dans le mélange.
Pourquoi la première étape de la revue littéraire? Permettez-moi de vous rappeler que nous n'avons pas de médicament, mais un appareil médical. Il n'est pas nécessaire de tester la nouvelle formule. Plus souvent qu'autrement.
Cela ressemble à ceci: nous nous référons à des articles pour examen, c'est-à-dire que nous trouvons des magazines dans les archives, nous notarions des captures d'écran et des copies afin que la commission elle-même ne regarde pas plus tard.
Ensuite, nous avons besoin de la rétroaction de la pratique clinique sur les analogues enregistrés. Autrement dit, nous devons trouver des publications (ou prendre l'analogue le plus proche déjà enregistré une fois et vérifier comment cela résout ce problème). Sur la base des informations collectées sur les analogues enregistrés, un acte d'évaluation des résultats des essais cliniques de dispositifs médicaux est élaboré.
Ensuite, avec un ensemble de documents confirmant l'achèvement de toutes les étapes indiquées ci-dessus, ainsi que d'autres documents supplémentaires pour le dispositif médical, nous sommes envoyés à Roszdravnadzor pour enregistrement.
Comment se déroule le processus?
Tout d'abord, une demande et un dossier pour le futur produit sont soumis à Roszdravnadzor. L'application décrit l'objectif, la classe de risque, la portée et d'autres informations sur le dispositif médical, ainsi que des informations sur le fabricant. En plus de l'application, un ensemble de documents est joint: protocoles, certificats de tous les tests effectués, documentation technique (TU), documentation opérationnelle (mode d'emploi, étiquettes), images photographiques, rapport d'analyse des risques, informations sur les documents réglementaires, certificat de test de qualification, et contrats de location et autres documents du fabricant.
C'est-à-dire qu'au moment de la soumission de la demande, il est nécessaire de terminer tous les tests internes (dans notre production, dans notre propre laboratoire, ils sont rendus plus difficiles que dans les laboratoires d'État, car il est élémentaire plus rentable d'y aller dans un premier temps et de ne pas rencontrer de revues de parties plus tard dans le processus de vente). Passez ensuite la toxicologie à l'un des laboratoires accrédités par l'État.
Dans notre cas, par exemple,
Blefarogel (un moyen pour l'hygiène des paupières dans la prévention
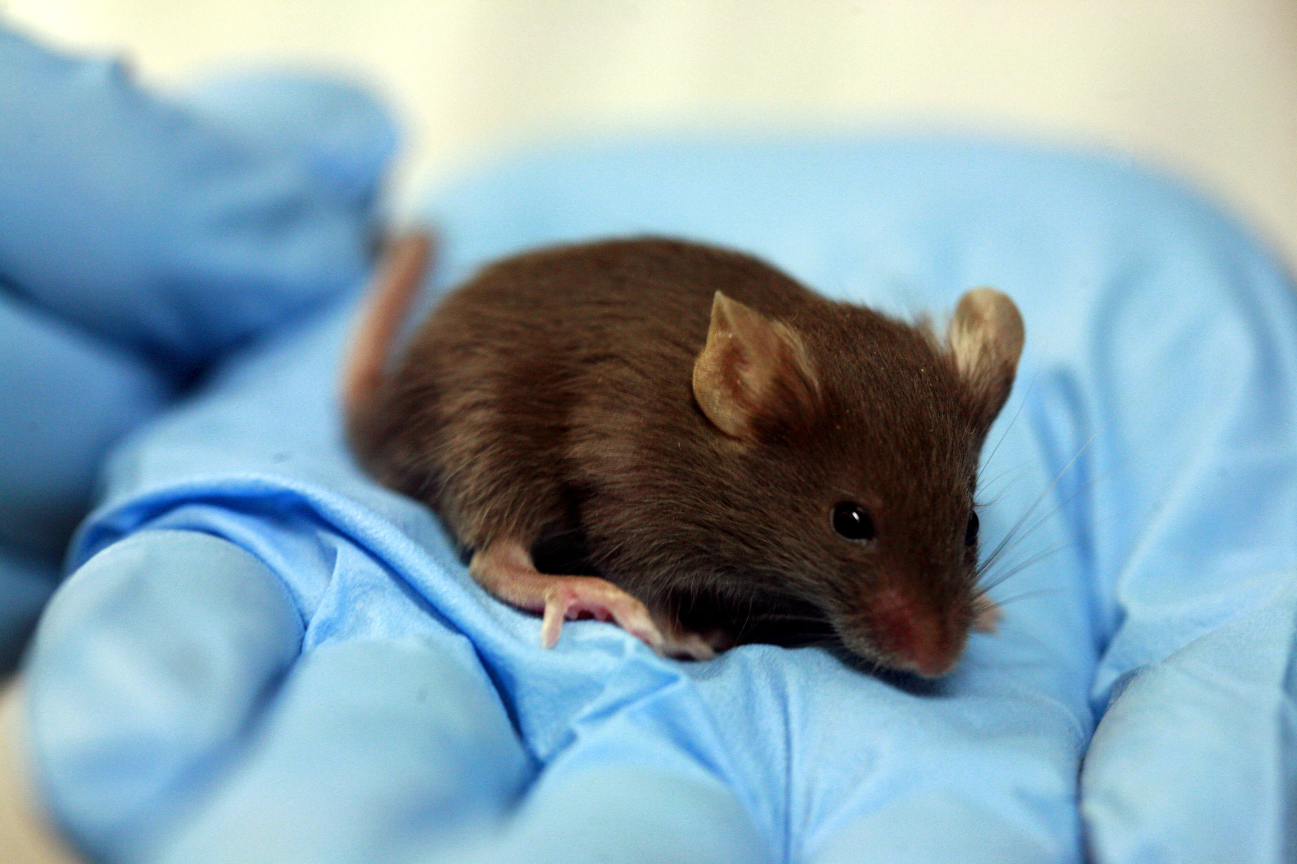
du syndrome de l'œil sec ) est un gel qui est appliqué sur les paupières et la peau du visage. La toxicologie était des souris. Je ne sais pas exactement ce qu’ils ont fait avec eux, mais je suppose qu’ils ont rasé deux zones sur chaque animal, l’une enduite de gel, la seconde à gauche ou enduite de quelque chose de neutre. Et regardé la souris. Heureusement, ils n'ont pas poussé à l'intérieur de la souris. (D'accord, ils ne l'ont presque pas poussé - le fait est que dans la fiche de données de sécurité [ou MSDS - fiche de données de sécurité] du propylène glycol, il y a des données sur la dose létale lorsqu'il est pris par voie orale. Il a besoin de 4 grammes par souris [il se casse physiquement en même temps le plus probable] Et après tout, ils ont en quelque sorte fixé cette dose ...) Mais revenons aux produits de souris les plus réussis, qui sont tout simplement barbouillés. Ils ont des voisins qui ont obtenu des bouteilles propres du produit, dans lesquelles ils versent de l'eau et se défendent. Ensuite, ils sont enduits de cette eau des bouteilles pour vérifier que le plastique ne libère pas quelque chose de dangereux au fil du temps. Ou non: la méthodologie exacte n'a pas été divulguée aux fabricants, il y a une forte probabilité que cette eau de la bouteille soit simplement analysée chimiquement et, même en cas de suspicion, injectée par voie intradermique.
Nous sommes désolés pour les souris, mais l'approche est la suivante: sans nos tests, elles ne seraient pas nées du tout. En général, ils n'y sont pas tourmentés (au moins, nous arrivons déjà avec une étude toute prête sur nous-mêmes). Nous savons également que les assistants de laboratoire éprouvent des sentiments chaleureux pour les souris, comparables à l’amour des villageois pour le poulet. Et nous soupçonnons qu'à la fin de la recherche, la souris n'est pas complètement éliminée, mais est envoyée en rééducation avant de nouveaux tests. Mais ce n'est qu'une théorie: s'il y a quelqu'un ici qui peut parler de son destin, ce serait merveilleux.
La même demande doit être accompagnée d'une recherche technique. Plus précisément, rédigez le TU (document réglementaire pour un dispositif médical qui établit des exigences techniques) et prouvez que le dispositif médical y correspond. Les experts s'entendent sur nos conditions. Ensuite, la stabilité des échantillons en série et la conformité de leurs dossiers produits y sont vérifiées: couleur, odeur, conductivité électrique, atténuation du signal ultrasonore, etc. - tout cela est indiqué. À ce stade, il est souvent nécessaire de reformuler un mot particulier dans la description du produit. Notre Mediagel a une viscosité différente: il s'agit d'un certificat d'enregistrement, mais trois kits de test sont pour une viscosité moyenne, un gel plus liquide et plus dense.
Nous effectuons les mêmes contrôles réguliers de conformité aux TU dans notre propre production. Ce n'est qu'une fois dans l'entraînement qu'il y a eu un cas où nous nous sommes approchés des limites admissibles du test, généralement nous marchons exactement au centre des tolérances entre les limites.
La classe de risque du produit est déterminée par son objectif, sa méthode d'utilisation, sa durée d'utilisation, son caractère invasif et d'autres critères. Nous avons des gels à ultrasons, par exemple, la durée de contact avec la peau jusqu'à une heure est la première classe. Et dans la deuxième classe, par exemple, les miroirs gynécologiques, différentes choses pour les nouveau-nés tombent.
Il y a une différence dans les essais cliniques. Première classe - les essais cliniques peuvent être effectués immédiatement. La deuxième classe et au-dessus - la clinique seulement après l'autorisation officielle de Roszdravnadzor. Avant les essais cliniques, l'équipe de développement n'a le droit de tester que sur elle-même. Le but est de vérifier l'effet principal. L'objectif de la bêta est de comprendre les effets secondaires possibles et divers cas intéressants, tels que «tout en prenant d'autres médicaments». Les essais cliniques commencent lorsque la sécurité du dispositif médical est prouvée en théorie.
Je constate que s'il ne s'agit pas de produits médicaux, mais de cosmétiques, alors des études sont menées sur des volontaires dans des cliniques accréditées pour des recherches scientifiques de ce type.
En pratique, ceci est le suivant: la commission achève l'analyse des documents (à l'exception des dispositifs médicaux de 1ère classe de risque et des dispositifs médicaux de diagnostic in vitro), puis elle envoie une lettre et donne 50 jours ouvrables pour mener des études cliniques et leur communiquer les résultats. Pour la plupart de nos gels, toutes les pratiques cliniques ont été obtenues avant même le resserrement des exigences, il n'y a donc pas eu de difficultés. Mais maintenant, nous avons une commande cible d'ophtalmologistes pour le nouveau Blefarogel Forte - c'est comme le Blefarogel-2 avec des composés soufrés pour le démodex, uniquement pour les cas avancés. Si Blefarogel-1 est «pour que mes yeux ne me blessent pas lorsque je suis assis devant l'ordinateur», Blefarogel-2 est «pour qu'il n'y ait pas d'inflammation de la tique et qu'il n'ait rien à manger pour toujours», Blefarogel-Forte sera «génocide des tiques à tout prix». En ce sens que nous augmentons délibérément le risque d'allergies en augmentant la proportion de substances actives, mais nous recombinons la formule pour qu'elle aide dans les cas particulièrement difficiles. Car cela se fait à la demande de médecins qui anticipent seulement avoir besoin d'un tel outil.
Fin de l'épopée
Ensuite, l'exhaustivité des documents est vérifiée à Roszdravnadzor: un consultant exécutif qui vérifie simplement le colis se démarque. Les documents sont numérisés et envoyés à une organisation experte du département de Roszdravnadzor. En conséquence, une conclusion est établie à Roszdrav sur la base de la totalité de tous les documents. En fait un groupe d'experts. Si tout va bien et que les documents correspondent les uns aux autres, ils autorisent l'enregistrement de l'État.
À partir de ce moment, vous ne pouvez plus rien toucher. Remplacer la lettre sur l'étiquette ou remplacer l'icône signifie automatiquement que vous devez vous réinscrire. On nous a demandé de changer l'étiquette de Mediagel-s en russe-anglais. Cela signifie un nouvel enregistrement. Il a fallu six mois pour saisir le texte anglais.
Le processus étant expert, il est largement associé à des interprétations spécifiques de normes assez complexes. Cela signifie que plusieurs fois, vous pouvez soumettre des documents selon le même schéma, puis il s'avère qu'un nouveau morceau de papier sera nécessaire. En général, il s'agit d'une sorte d'accident: ils peuvent se terminer à chaque nouvelle soumission. Dans le même temps, l'ancien enregistrement d'État demeure et ne disparaît pas, mais nous perdons de l'argent pour les procès et payons un droit d'État important, même pour l'enregistrement et les modifications. Et nous dépensons des souris d'État.
Ensuite, vous devez attendre la commande sur papier. Lorsqu'il y a une commande, le produit ira au registre, c'est-à-dire au
site sur lequel les pharmacies regardent ce qui est quoi. Ce n'est qu'à partir de ce moment que vous pouvez faire quelque chose. La pharmacie ne prend pas de produits qui ne sont pas dans l'EF du Ministère de la Santé, si ce n'est pas des cosmétiques bien sûr.
Ce n'est qu'à ce moment que la production peut commencer. Sans le numéro du ministère de la Santé, nous pouvons non seulement vendre, mais même produire en Russie.
Tout, à partir de ce moment, nous fabriquons le dispositif médical en série. Par expérience: il est très important d'avoir un laboratoire de contrôle de la qualité bien développé, car toute plainte signifie un contrôle approfondi à l'usine (à en juger par le comportement, vous ne pouvez pas revenir sans rapport d'au moins quelques violations) et arrêter les expéditions. À cet égard, nous préférons effectuer nous-mêmes des tests d'acceptation et des tests périodiques et clore les études sur de petits échantillons avec les nôtres. Je veux dire que 10 volontaires de la norme sont peu nombreux, mais 100 volontaires sont déjà un échantillon plus ou moins représentatif. À long terme, des recherches plus approfondies sont toujours payantes, et pour moi, c'est un grand mystère pourquoi les autres fabricants ne le font pas toujours.
Dans le prochain article, je vais essayer de parler d'une autre étape importante de la recherche - le test de provocation: c'est à ce moment que les échantillons sont spécialement contaminés par des bactéries pathogènes. Cette partie de la recherche ne doit jamais être effectuée à proximité de laboratoires de production ou de recherche, de sorte que les tests de provocation sont effectués.