Mitochondries - petits travailleurs ou grands patrons?Si vous pensez que l'histoire la plus importante pour nous de vivre ensemble commence pendant le mariage, alors ce n'est pas du tout le cas. L'histoire la plus importante de la vie de chaque personne a commencé il y a plus d'un milliard d'années lorsque nos lointains ancêtres unicellulaires ont été contraints de signer un «contrat de mariage» avec ceux que nous appelons maintenant les mitochondries (voir la théorie de la symbiogenèse).
Les mitochondries ont deux membranes (interne et externe) et leur propre matériel héréditaire sous forme d'ADN (Fig.1). Sur la membrane interne des mitochondries, il existe un système de phosphorylation oxydative, dont le fonctionnement assure l'oxydation des substrats énergétiques avec la formation d'ATP.
Fig. 1 . Structure schématique des mitochondries
Dans le contrat de mariage de la cellule et des mitochondries, il n'y a pas de clause "dans la maladie et la santé" - et bonne. Si les mitochondries vieillissent, la cellule peut la tuer pendant la mitophagie, et les mitochondries, à leur tour, régulent le processus d'apoptose dans les cellules dysfonctionnelles et anciennes. Si le processus de contrôle mutuel de la qualité est perturbé, des mécanismes de vieillissement sont lancés. Les mécanismes de l'apoptose sont perturbés, le nombre de radicaux libres non contrôlés par les mitochondries augmente. Cela provoque une inflammation systémique, des dommages à l'ADN de la cellule. Ainsi, il existe une relation étroite entre la dysfonction MX, les maladies liées à l'âge, le vieillissement et les dysfonctionnements métaboliques [1]. La dysfonction métabolique est un rider constant de l'apocalypse du vieillissement.
"Comme un écureuil dans une roue" - la dynamique des mitochondries
Tout le blâme pour les troubles métaboliques ne réside pas dans notre suralimentation. Les troubles métaboliques sont principalement associés à l'incapacité des mitochondries à faire face aux nutriments. Les mitochondries dans la cellule ne sont pas faciles. Nous «nourrissons» nos cellules soit trop, soit trop peu, et leur présentons une «demande» de donner de l'énergie sous forme d'ATP, dont la quantité doit correspondre exactement à nos besoins. Afin de «sortir» régulièrement de cette situation, les mitochondries utilisent réellement certains «mouvements» - fission et fusion. Ces «mitodomotion» sont réunies sous le nom de «dynamique mitochondriale». L'équilibre entre la division mitochondriale et la fusion est le mécanisme central de l'adaptation bioénergétique aux besoins métaboliques de la cellule [2, 3].
La plupart des mitochondries se trouvent dans les tissus ayant des besoins énergétiques élevés - les muscles, le foie, le tissu adipeux brun et le cerveau. Il n'est pas surprenant que la dynamique des mitochondries dans ces tissus ait été mieux étudiée.
Donc, si une cellule de l'un de ces tissus (à l'exception de certains neurones du cerveau, plus sur cela plus tard) reçoit une grande quantité de nutriments (l'apport dépasse le coût), alors les mitochondries sont dans un état divisé (fragmenté). Si la cellule est dans un état de faim (revenu inférieur au coût), les mitochondries fusionnent et elles sont dans un état connecté. [3,4]. C'est ainsi que l'homéostasie cellulaire est maintenue (Fig. 2).
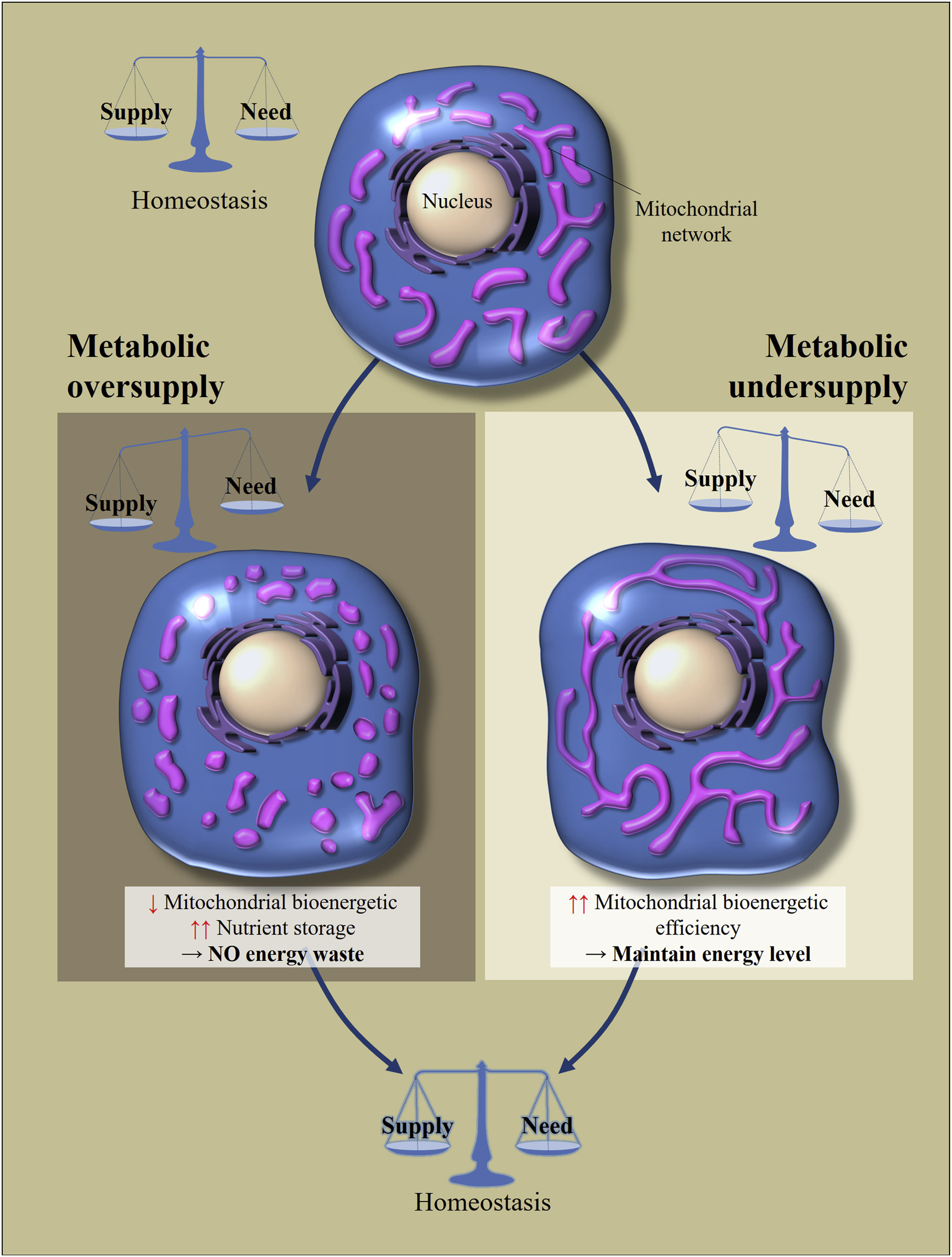 Fig. 2
Fig. 2 Régulation de la morphologie et de l'efficacité bioénergétique des mitochondries en réponse à un apport excessif ou insuffisant de nutriments [sur 2]
L'homéostasie métabolique cellulaire dépend de l'équilibre entre l'apport en nutriments et sa consommation. Les changements dans l'apport de nutriments entraînent des adaptations cellulaires pour rétablir l'équilibre. Un excès de nutrition entraîne une fragmentation du réseau mitochondrial, ce qui entraîne une diminution de l'efficacité bioénergétique des mitochondries. Cela évitera une perte d'énergie. En revanche, avec la faim métabolique, les mitochondries s'allongent afin d'augmenter leur efficacité bioénergétique.Quelle est l'astuce de ces mouvements? Si la cellule est en état de faim, la fusion des mitochondries peut augmenter leur efficacité bioénergétique (la quantité d'ATP créée par molécule nutritive). Si un excès de nutriments pénètre dans la cellule, ils peuvent soit 1) être stockés, soit 2) dissiper cette énergie sous forme de chaleur. La tâche des mitochondries dans ce cas est de dissiper plus d'énergie sous forme de chaleur, de stocker moins sous forme d'ATP (l'accumulation de NADH et de ROS entraînera un stress oxydatif). La fragmentation des mitochondries leur permet de réduire l'efficacité de la bioénergie, dont le principal mécanisme de réduction est considéré comme une «fuite» de protons.
Nous allons donc au travail, et la vie des mitochondries se déroule constamment dans le cycle de division et de fusion (Fig. 3).
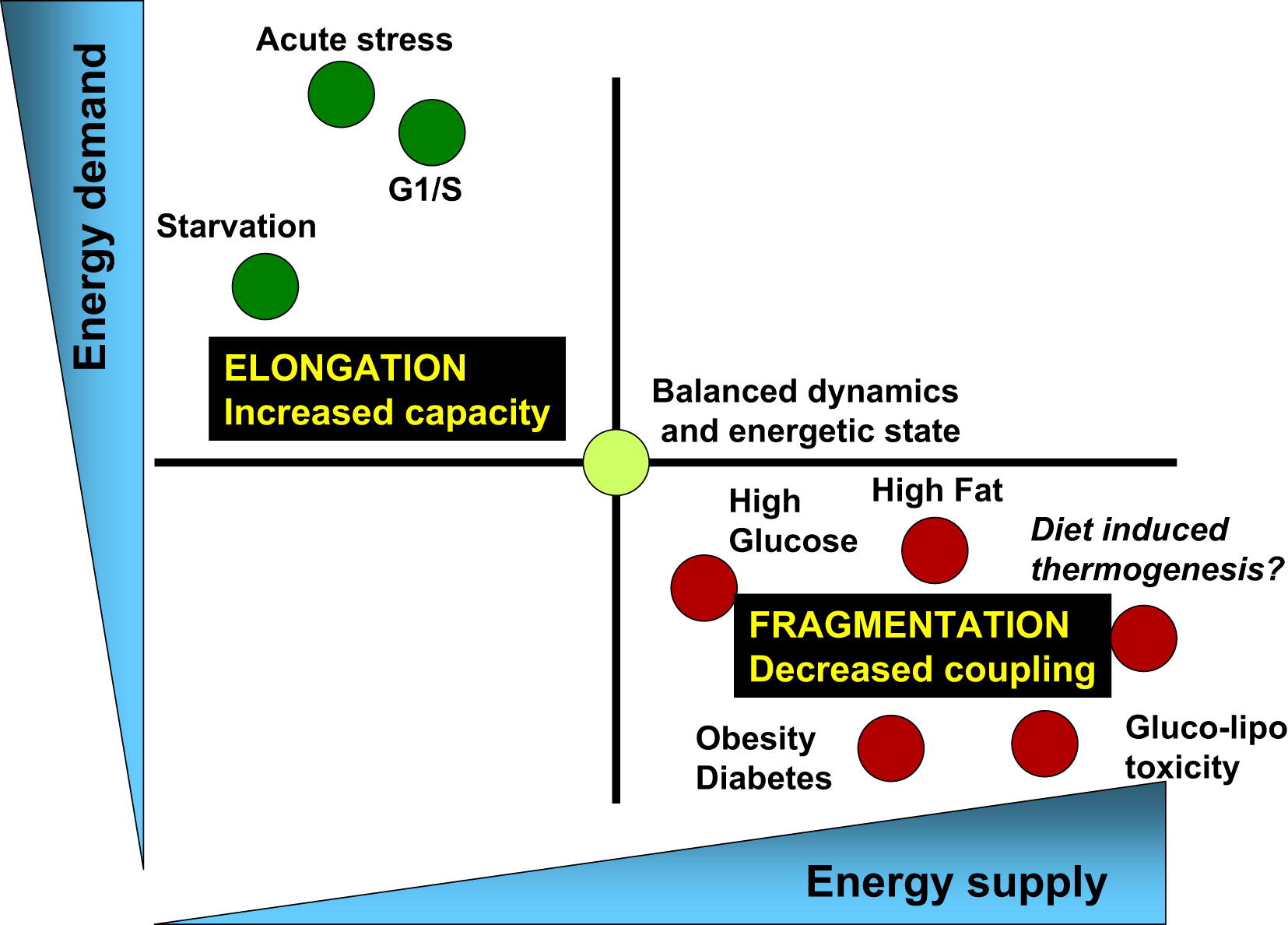 Fig. 3 L' équilibre de la consommation d'énergie et de l'approvisionnement énergétique est associé aux changements correspondants dans l'architecture des mitochondries et leur efficacité bioénergétique [sur 3]
Fig. 3 L' équilibre de la consommation d'énergie et de l'approvisionnement énergétique est associé aux changements correspondants dans l'architecture des mitochondries et leur efficacité bioénergétique [sur 3]
Les processus physiologiques associés à une augmentation de la demande d'énergie et à une diminution de l'approvisionnement énergétique (par exemple, stress aigu, famine et phase G1 / S) sont caractérisés par un allongement des mitochondries et une respiration associés à la synthèse d'ATP. En revanche, les processus physiologiques associés à une diminution de la demande énergétique et à une augmentation de son approvisionnement (niveaux élevés de nutriments, obésité et diabète de type 2) sont associés à la fragmentation des mitochondries, à la production de chaleur ou à une diminution de la fonction mitochondriale.Des cycles de fission et de fusion sains sont essentiels à la santé métabolique des cellules
Le cycle normal de division et de fusion mitochondriales est un élément clé de leur contrôle qualité. Pourquoi? Pendant la division mitochondriale, deux filles se forment, dont l'une a un potentiel de membrane plus élevé et va plus loin dans le cycle de fusion-division, et l'autre, avec une membrane plus dépolarisée, reste séparée jusqu'à ce que le potentiel de membrane soit restauré. Si le potentiel est restauré, il retrouve le réseau mitochondrial. S'il reste dépolarisé, il est alors éliminé en cours d'autophagie, qui est la clé de la qualité du pool mitochondrial (Fig.4).
L'inhibition à long terme de la division mitochondriale (avec une famine cellulaire prolongée) conduit à l'accumulation de mitochondries endommagées, qui ne peuvent pas être séparées [3, 4].
D'un autre côté, un excès de nutriments conduit à l'inhibition de la fusion mitochondriale, ce qui entraîne une perturbation du cycle de la dynamique mitochondriale, augmente l'hétérogénéité mitochondriale intracellulaire. Oui, avec un excès de nourriture, la fragmentation mitochondriale est protectrice, mais une fragmentation prolongée, comme une fusion prolongée, est nocive pour le contrôle de la qualité mitochondriale. Il n'y a pas d'élimination sélective; la masse mitochondriale diminuera et sera constituée de petites mitochondries dépolarisées.
 Fig. 4 Cycle de vie des mitochondries et sa régulation de la disponibilité des nutriments [sur 3]
Fig. 4 Cycle de vie des mitochondries et sa régulation de la disponibilité des nutriments [sur 3]Les mitofusines ne sont pas seulement des protéines
Au niveau moléculaire, la fusion mitochondriale est un processus en deux étapes qui nécessite une fusion coordonnée des membranes externe et interne lors d'événements séquentiels séparés. Chez les mammifères, ce processus est régulé par trois protéines qui appartiennent aux GTPases: Mfn1 et Mfn2 sont nécessaires pour la fusion de la membrane externe, et OPA1 - pour la fusion de la membrane interne. D'autres protéines sont nécessaires pour la division, Fis1 et Drp1.
Le rôle des protéines de mitofusine a été étudié dans les études de perte et de gain de fonction. Des souris mutantes pour les protéines de mitofusine meurent dès la mi-gestation, car la fusion mitochondriale leur devient impossible. Les mitofusines sont importantes pour les processus d'autophagie et de mitophagie. Une diminution de l'expression de Mfn2 dans les cardiomyocytes bloque le début du processus d'autophagie, car la fusion des autophagosomes avec les lysosomes est bloquée. L'épuisement de Mfn2 entraîne une diminution du potentiel des membranes mitochondriales, pour compenser, il y a une diminution du travail de la chaîne respiratoire, l'augmentation de l'absorption du glucose et la synthèse du glycogène diminuent. La cellule passe à la glyocyse anaérobie, et c'est la voie vers la dégénérescence oncologique de la cellule. La carence en Mfn2 entraîne des modifications neurodégénératives. Une augmentation de l'expression de Mfn2 dans les muscles squelettiques augmente leur sensibilité à l'insuline.
Mfn1 remplit des fonctions similaires, mais probablement dans d'autres tissus (l'expression de Mfn2 et Mfn1 varie selon les tissus) - Mfn1 s'exprime davantage dans le cœur, le foie, le pancréas, les testicules et Mfn2 dans le cœur, les muscles squelettiques, le cerveau, le tissu adipeux brun .
Ainsi, les mitofusines sont des régulateurs clés de la dynamique mitochondriale. L'expression des mitofusines est différente dans les différents organes, elles fournissent l'efficacité bioénergétique et les mécanismes d'adaptation à la disponibilité des nutriments, et le "destin" de la cellule en dépend. Sans surprise, les protéines de fusion mitochondriales sont des cibles potentielles pour des interventions pharmacologiques [2, 5].
Hypothalamus, mitochondries, dysfonctionnement métabolique et vieillissement
La dynamique des mitochondries est importante dans toutes les cellules. Dans les cellules bêta du pancréas, les mitochondries sont des capteurs de nutriments et des générateurs de signaux de synthèse d'insuline, dans les muscles, la dynamique mitochondriale est importante pour la régulation du métabolisme du glucose, etc. Cependant, une personne n'est pas seulement un ensemble de différents types de cellules, chacune prenant des décisions indépendantes. Un organisme est un système qui a un lien régulateur central pour maintenir l'énergie et l'homéostasie du glucose. Ce principal régulateur est l'hypothalamus.
L'hypothalamus est situé dans le diencéphale et c'est lui qui assure l'interconnexion des systèmes de régulation nerveux et humoral. Les neurones hypothalamiques perçoivent, traitent et répondent aux signaux du tissu adipeux (leptine), du pancréas (insuline) et d'autres stimuli hormonaux (ghréline, cholécystokinine, polypeptide pancréatique, etc.). L'hypothalamus contrôle l'activité du système endocrinien humain du fait que ses neurones sont capables de sécréter des émetteurs neuroendocriniens qui stimulent ou inhibent la production d'hormones par l'hypophyse. En d'autres termes, l'hypothalamus, dont la masse ne dépasse pas 5% du cerveau, est le centre de régulation des fonctions endocriniennes et de maintien de l'homéostasie de l'organisme tout entier.
Même Dilman (Dilman V. M "Grande horloge biologique") a souligné le rôle moteur de l'hypothalamus dans le développement systématique de la dysfonction métabolique, conduisant à l'obésité, au diabète, aux maladies cardiovasculaires, oncologiques et au vieillissement. Selon la théorie de l'hyperadaptose formée par Dilman, la sensibilité des récepteurs hypothalamiques aux signaux provenant des tissus corporels (leptine, insuline, etc.) diminue progressivement systématiquement avec l'âge. Pour provoquer sa «réponse», il faut de plus en plus de l'une ou l'autre hormone - plus d'insuline, plus de leptine. Développe la résistance à l'insuline et à la leptine, les maladies métaboliques entraînant le vieillissement et la mort.
Selon les fonctions exercées, des groupes de neurones sont combinés dans les noyaux de l'hypothalamus. L'un d'eux - le noyau arqué (arqué) est un régulateur clé du comportement alimentaire et du métabolisme. Des neuropeptides orrexigènes (stimulent l'appétit) et anorexigènes (suppriment l'appétit), correspondant respectivement aux neurones AgRP et POMC, peuvent s'y former. Les signaux périphériques (insuline, ghréline, leptine, etc.) affectent l'expression des peptides qui stimulent ou suppriment l'appétit, ce qui assure la cohérence de la régulation centrale (Fig. 5).
 Fig. 5. Contrôle hypothalamique du métabolisme énergétique. Le cerveau intègre les signaux métaboliques (leptine, insuline, ghréline, PYY3-36) des tissus périphériques tels que le pancréas, le tissu adipeux et l'estomac. Dans le cerveau, des réseaux neuronaux spécialisés coordonnent les changements adaptatifs dans l'absorption et la consommation de nourriture [sur 5].
Fig. 5. Contrôle hypothalamique du métabolisme énergétique. Le cerveau intègre les signaux métaboliques (leptine, insuline, ghréline, PYY3-36) des tissus périphériques tels que le pancréas, le tissu adipeux et l'estomac. Dans le cerveau, des réseaux neuronaux spécialisés coordonnent les changements adaptatifs dans l'absorption et la consommation de nourriture [sur 5].Alors, qui et comment régule la sensibilité des neurones hypothalamiques?
Une étude de la dynamique des mitochondries dans les tissus cérébraux a montré que la dynamique des mitochondries joue un rôle important dans la capacité des neurones hypothalamiques à contrôler le taux de glucose et l'homéostasie énergétique dans le corps [6,7,8].
Dans les neurones AgRP (neurones AgRP favorisant la faim), qui stimulent l'appétit et régulent la prise de poids, la famine entraîne une division mitochondriale et une alimentation riche en graisses conduit à la fusion. Autrement dit, la réponse des mitochondries est différente de celle de la plupart des autres cellules.
La fusion de MX dans ces neurones régule l'activité électrique en réponse à un régime riche en graisses, stimulant la production d'un peptide orexigène (peptide AgRP), qui est nécessaire pour la prise de poids et le dépôt de graisse avec un excès de nutriments. Les suppressions de Mfn1 et Mfn2 dans ces neurones ont entraîné une diminution du gain de poids chez les rats en raison de niveaux plus faibles de leptine circulante.
Les neurones POMC (suppriment l'appétit) ont la fonction opposée, et la dynamique des mitochondries en réponse à l'apport en nutriments est différente. Une diminution de l'expression des mitofusines dans ces neurones entraîne une perturbation de la connexion des mitochondries avec l'EPS et, par conséquent, l'hyperphagie, la résistance à la leptine et l'obésité. Dans le même temps, la consommation alimentaire a augmenté et la consommation d'énergie a diminué.
Ainsi, la réponse du corps à un régime riche en graisses dépend des modèles de dynamique mitochondriale dans les neurones hypothalamiques. Le remodelage des mitochondries dans les neurones assure leur réponse à l'apport en nutriments, stimule la production de neuropeptides qui stimuleront ou supprimeront l'appétit, affectant le métabolisme au niveau du corps (Fig.6).
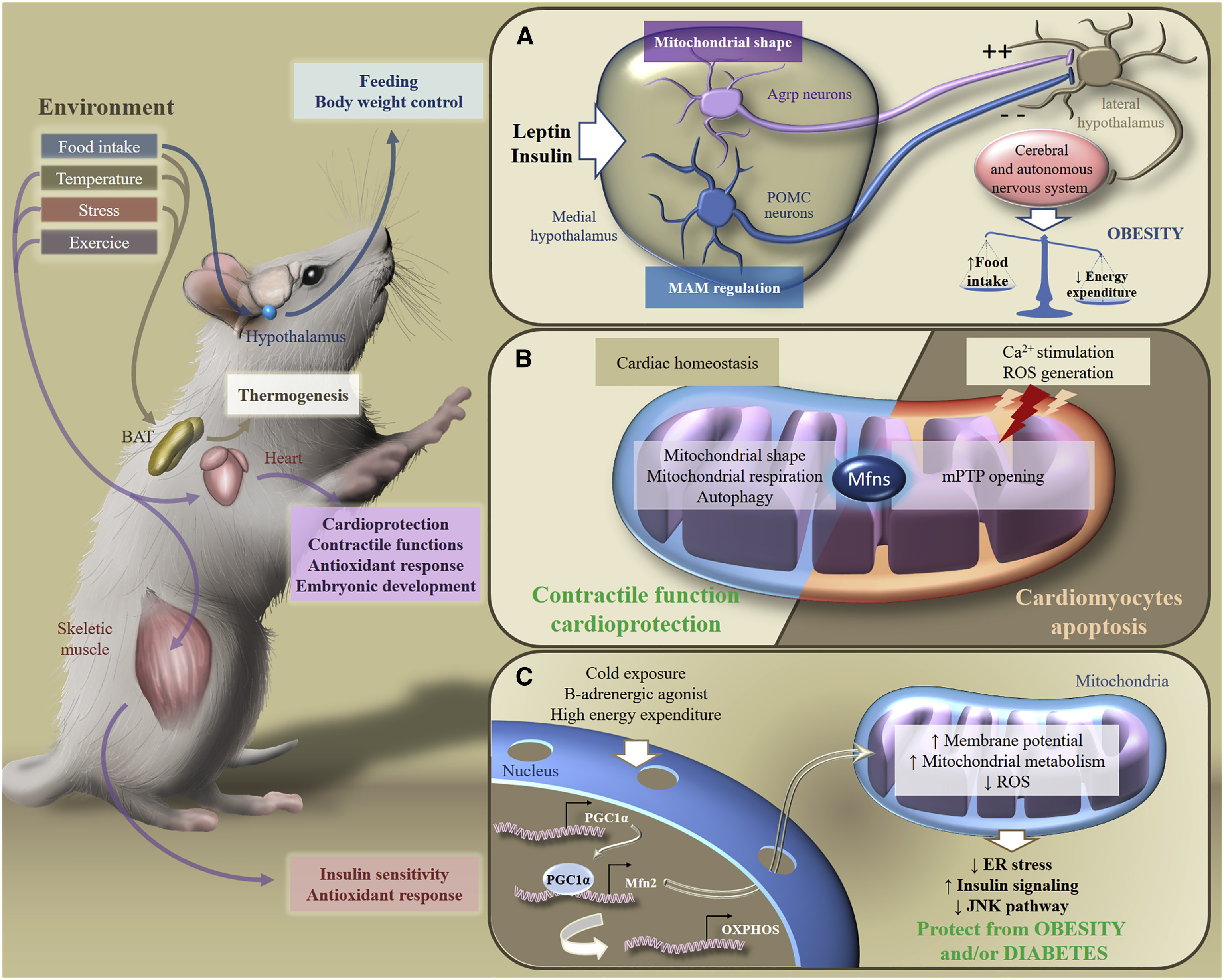 Fig.6. Adaptation métabolique aux stimuli environnementaux [de 2]
Fig.6. Adaptation métabolique aux stimuli environnementaux [de 2]En réponse à des stimuli exogènes, les Mfns participent à la transduction de la signalisation métabolique dans divers organes, ce qui assure le maintien de l'homéostasie énergétique dans tout le corps. En particulier, en réponse à l'apport alimentaire, aux changements de température, au stress ou à l'exercice, le tissu adipeux brun, le cerveau, le cœur ou les muscles squelettiques adaptent leur métabolisme pour contrôler la nutrition, le poids corporel, la fonction contractile, la réponse antioxydante ou la sensibilité à l'insuline.
Comment influencer la dynamique des mitochondries?
1. Nutrition et exerciceCycles de nutrition Un excès de nourriture et un régime riche en graisses (HFD) inhibent la fusion mitochondriale dans les cellules (le mécanisme est différent dans certains neurones cérébraux). Un cycle incomplet de division-fusion mitochondriale perturbe les processus d'autophagie → l'augmentation de l'hétérogénéité mitochondriale intracellulaire → il n'y a pas d'élimination sélective des mitochondries → les mitochondries avec dysfonctionnement s'accumulent.
La restriction calorique (cycle alimentation / jeûne) stimule l'adaptation bioénergétique, fournissant des mécanismes de qualité mitochondriale.
2. Membranes saines: acide stéarique, cardiolipine, acide phosphatidiqueTous les processus clés dépendent de la «santé» des membranes mitochondriales - autophagie, mitophagie, apoptose, la relation des mitochondries avec le réticulum endoplasmique et la dynamique des mitochondries. Les membranes des organites cellulaires sont composées de lipides et de protéines. Le remodelage de ces membranes est contrôlé par des interactions entre des lipides et des protéines spécifiques.
Les acides gras saturés comprennent la palmitique (C16) et la stéarique (C18). Il a été démontré que l'utilisation d'acide stéarique (C18: 0) stimule le processus de fusion mitochondriale. Son action est associée à un effet sur les mitofusines. Chez la souris, les compléments alimentaires d'acide stéarique peuvent restaurer partiellement le dysfonctionnement mitochondrial provoqué par des mutations dans les gènes Pink1 ou parkin. Chez les neutrophiles de personnes qui ont suivi un régime à faible teneur en C18: 0 pendant 2 jours, les mitochondries sont dans un état fragmenté (50% des cellules avaient un MX fragmenté, 10% étaient liées au MX). L'utilisation d'acide stéarique les a amenés à fusionner les mitochondries après 3 heures [8]. Ainsi, l'acide stérique est important pour maintenir la dynamique mitochondriale. La plupart de l'acide stéarique se trouve dans les fèves de cacao (31-34%).
Les phospholipides sont les principaux composants des membranes organites. Ils régulent également la dynamique des mitochondries, et leur effet est différent [9].
La cardiolipine (CL) stimule la division mitochondriale et la fusion des membranes internes.
La cardiolipine est nécessaire au fonctionnement du complexe IV (citrochrome C oxydase) de la chaîne de transport d'électrons. La cardiolipine est située presque exclusivement dans la membrane interne des mitochondries. Avec l'âge, la quantité de cardiolipine diminue. Il existe une théorie selon laquelle la perte de la fonction cardiolipine est associée au remplacement des acides gras saturés dans sa molécule par des acides gras polyinsaturés. Pour résoudre ce problème, il est nécessaire d'introduire dans le régime des graisses saturées riches en acide gras stéarique tout d'abord.
Pour augmenter l'efficacité de la livraison des acides gras saturés à la membrane, des transporteurs peuvent être utilisés. Par exemple, l'utilisation de la phosphatidylcholine saturée (dipalmitophosphatidylcholine et dyseroylphosphatidylcholine), qui pourrait potentiellement délivrer des AG saturés directement à la cardiolipine [10]. La choline, en tant que support, passe facilement à travers le cytosol et pénètre dans les mitochondries.
L'acide phosphatidique (RA) inhibe la division mitochondriale et stimule la fusion des membranes externes (Fig. 7).
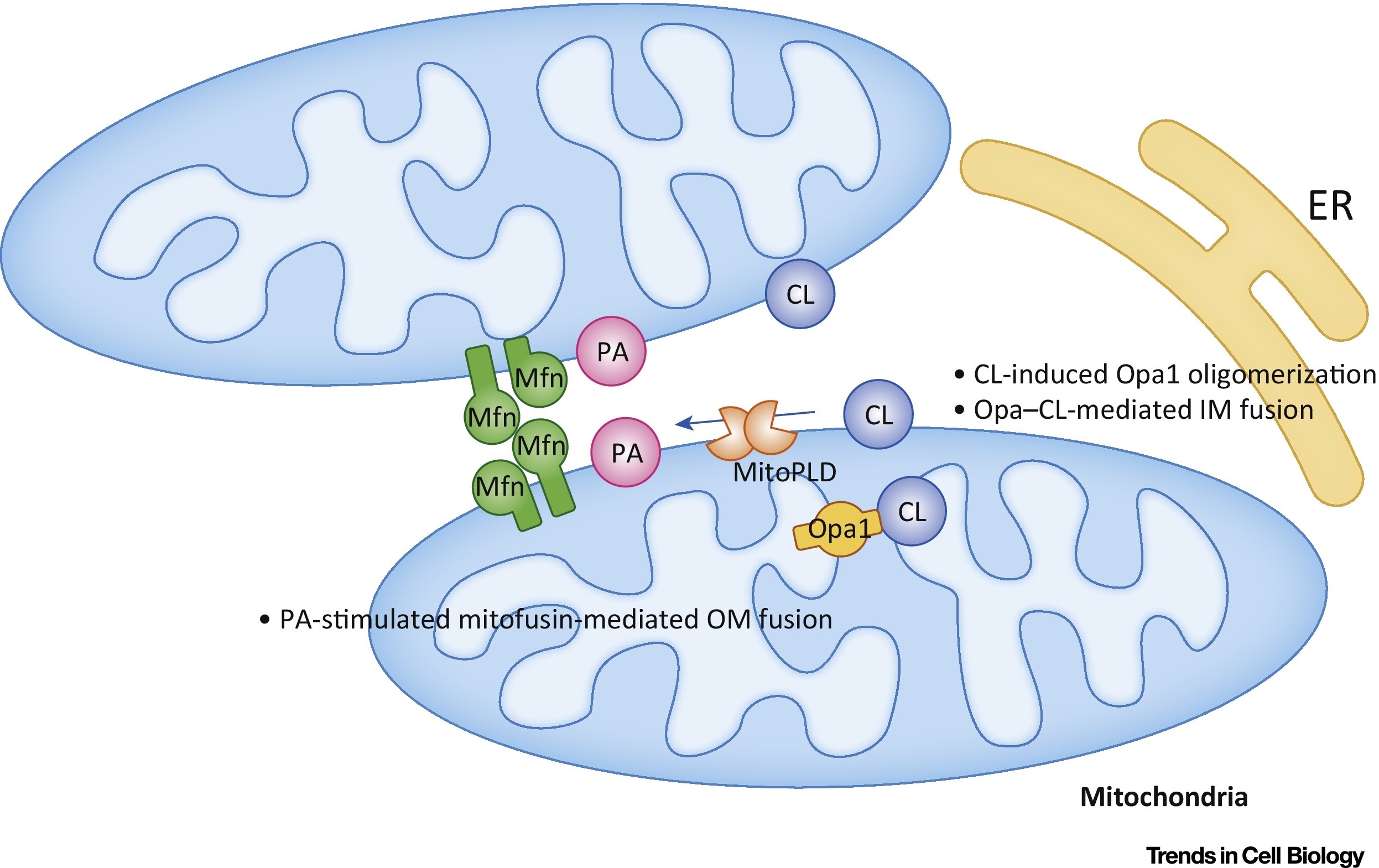 Fig. 7 Régulation de la fusion mitochondriale avec l'acide phosphatidique (PA) et la cardiolipine (CL) [sur 9].
Fig. 7 Régulation de la fusion mitochondriale avec l'acide phosphatidique (PA) et la cardiolipine (CL) [sur 9].Dans la membrane externe (OM), la PR stimule la fusion médiée par la mitofusine (Mfn). Dans la membrane interne (IM), CL stimule la fusion médiée par Opa1. Abréviations: ER - réticulum endoplasmique; MitoPLD, - phospholipase D. localisée dans les mitochondries
3. Régulation de l'expression des mitofusines (protéines responsables de la dynamique des mitochondries)Tout ce dont nous avons parlé plus haut (restriction calorique, acide stéarique, phospholipides) agit en affectant l'expression des mitofusines.De plus, il existe un certain nombre de médicaments qui peuvent indirectement affecter la dynamique des mitochondries. Il s'agit notamment de l'utilisation de la metformine.Le plus intéressant est l'utilisation de substances pouvant affecter directement l'expression des mitofusines. L'un des médicaments potentiels appelé léflunomide (léflunomide), qui a été approuvé par la FDA [5,11]. Il est un inducteur de l'expression de Mfn1 et Mfn2 et a été enregistré comme médicament pour le traitement de la polyarthrite rhumatoïde.Thérapie génique mitochondriale
Une dynamique mitochondriale altérée peut être associée à une altération de l'expression des protéines responsables de la fusion et de la division mitochondriales. De plus, le dysfonctionnement de ces protéines peut être associé (et cela arrive le plus souvent) à leurs mutations. Il existe deux approches pour l'examen des interactions de cause à effet du dysfonctionnement des mitochondries.On croyait auparavant que le mode de vie, y compris la suralimentation, conduit à la formation de radicaux libres, au stress oxydatif, à des mutations du génome mitochondrial et, par conséquent, à un dysfonctionnement des mitochondries. Cependant, il y a récemment des preuves convaincantes que les mutations de l'ADN mitochondrial sont inévitables, toutes ont des mutations ponctuelles de l'ADN hétéroplasmique et sont associées à des erreurs de réplication, et non à des dommages oxydatifs, auxquels l'ADN mitochondrial est assez stable [12]. Déjà au stade de l'œuf fécondé, certaines de nos mitochondries portent des mutations. Au fil du temps, ils se divisent, il y a plus de mitochondries mutantes, ils ne peuvent pas remplir leur fonction normalement.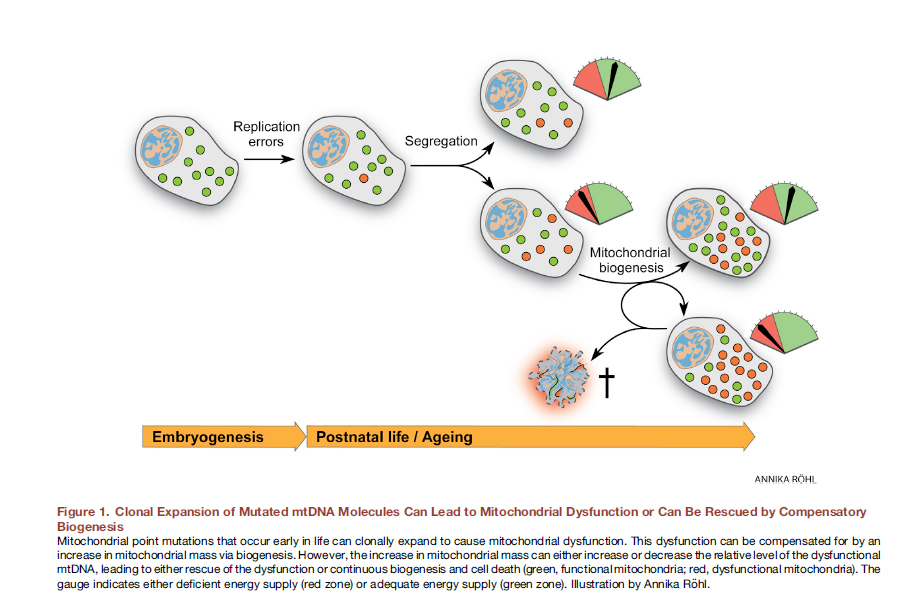 Fig. 8 L' expansion clonale des molécules d'ADNmt mutées peut entraîner un dysfonctionnement mitochondrial ou peut être «sauvée» par la biogenèse compensatoire [sur 12].Ici, in vivo, l'édition du génome mitochondrial pourrait être très utile. Il a été démontré que pour les mutations ponctuelles de l'ADN hétéroplasmique chez la souris, un succès significatif a déjà été obtenu avec des nucléases à doigts de zinc ciblées (mtZFN) avec livraison à l'aide d'un vecteur adénoviral [13].
Fig. 8 L' expansion clonale des molécules d'ADNmt mutées peut entraîner un dysfonctionnement mitochondrial ou peut être «sauvée» par la biogenèse compensatoire [sur 12].Ici, in vivo, l'édition du génome mitochondrial pourrait être très utile. Il a été démontré que pour les mutations ponctuelles de l'ADN hétéroplasmique chez la souris, un succès significatif a déjà été obtenu avec des nucléases à doigts de zinc ciblées (mtZFN) avec livraison à l'aide d'un vecteur adénoviral [13].Transfert mitochondrial
Une autre méthode prometteuse pour éliminer le dysfonctionnement mitochondrial est la transplantation mitochondriale. L'essence de cette approche est de «remplacer» les mitochondries endommagées par des mitochondries exogènes saines. Cette approche a d'abord été utilisée cliniquement chez les enfants atteints d'ischémie myocardique. Des mitochondries isolées autologues ont été utilisées pour la transplantation, qui ont été isolées avec le muscle droit de l'abdomen (une biopsie a été réalisée, puis la préparation a été préparée), puis administrée par injection directe [14]. Différentes approches pour l'introduction des mitochondries sont en cours de développement: injection directe de mitochondries isolées (injection locale) et injection systémique dans la circulation sanguine, lorsque les mitochondries elles-mêmes «recherchent» la cellule vers laquelle se rendre. Des groupes de chercheurs étudient la possibilité d'une transplantation mitochondriale dans la maladie de Parkinson, l'ischémie hépatique, les accidents vasculaires cérébraux, les maladies mitochondriales [15].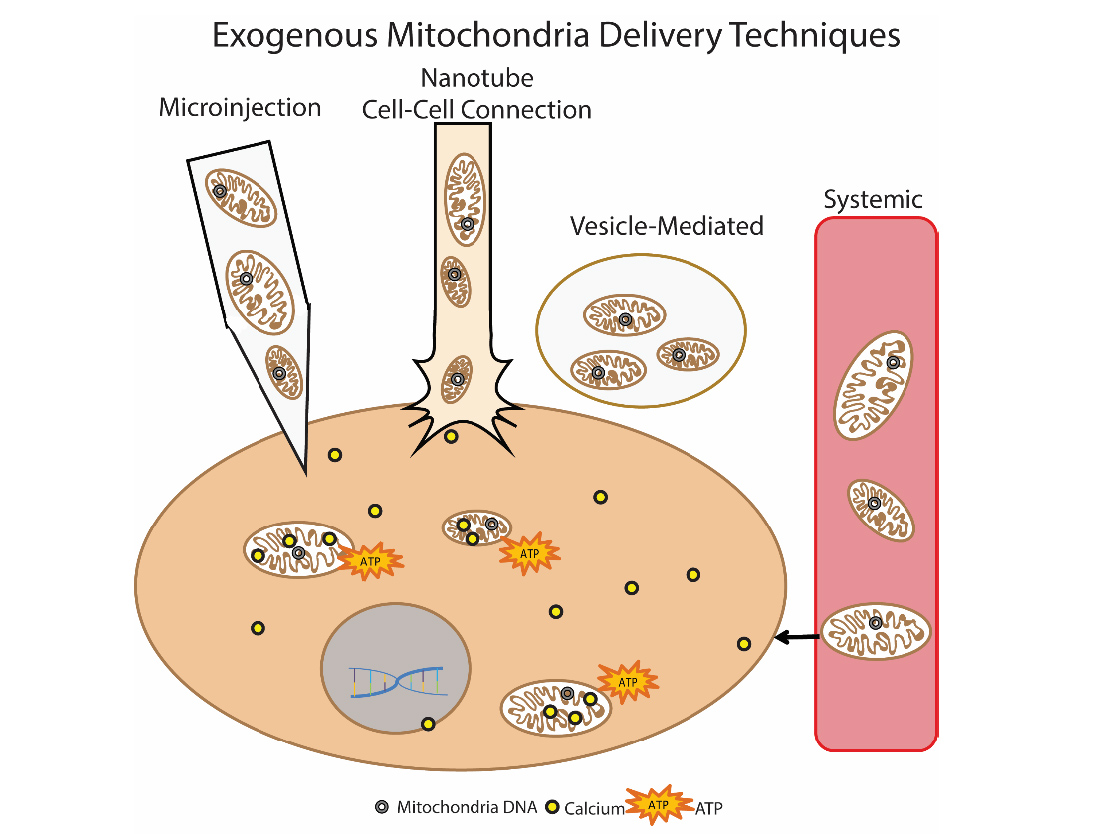 Fig. 9 Méthodes de délivrance des mitochondries exogènes à la celluleAuteur Olga Borisova
Fig. 9 Méthodes de délivrance des mitochondries exogènes à la celluleAuteur Olga BorisovaLittérature1. Kauppila, Timo ES, Johanna HK Kauppila, and Nils-Göran Larsson. «Mammalian mitochondria and aging: an update.» Cell metabolism 25.1 (2017): 57-71.
www.sciencedirect.com/science/article/pii/S15504131163050222. Schrepfer, Emilie, and Luca Scorrano. «Mitofusins, from mitochondria to metabolism.» Molecular cell 61.5 (2016): 683-694.
www.sciencedirect.com/science/article/pii/S1097276516001337#fig13. Marc Liesa, Orian Shirihai “Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure” Cell methabolism (2013): 491-506
www.sciencedirect.com/science/article/pii/S1550413113001046#fig34. Ramos, Eduardo Silva, Nils-Göran Larsson, and Arnaud Mourier. «Bioenergetic roles of mitochondrial fusion.» Biochimica et Biophysica Acta (BBA)-Bioenergetics 1857.8 (2016): 1277-1283.
www.sciencedirect.com/science/article/pii/S00052728163008585. Cunarro, Juan, et al. «Hypothalamic mitochondrial dysfunction as a target in obesity and metabolic disease.» Frontiers in endocrinology 9 (2018): 283.
www.frontiersin.org/articles/10.3389/fendo.2018.00283/full6. Marcelo O.Dietrich et al. «Mitochondrial Dynamics Controlled by Mitofusins Regulate Agrp Neuronal Activity and Diet-Induced Obesity”.
www.sciencedirect.com/science/article/pii/S0092867413010957#figs27. Steculorum, Sophie M., and Jens C. Brüning. „Sweet mitochondrial dynamics in VMH neurons.“ Cell metabolism 23.4 (2016): 577-579.
www.sciencedirect.com/science/article/pii/S15504131163011768. Senyilmaz-Tiebe, Deniz, et al. „Dietary stearic acid regulates mitochondria in vivo in humans.“ Nature communications 9.1 (2018): 3129.
www.nature.com/articles/s41467-018-05614-69. Kameoka, Shoichiro, et al. „Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics.“ Trends in cell biology (2017).
www.sciencedirect.com/science/article/pii/S096289241730158710.
raypeatforum.com/community/threads/mitolipin-liquid-saturated-phosphatidylcholine-pc-mix.1039811. Miret-Casals, Laia, et al. „Identification of new activators of mitochondrial fusion reveals a link between mitochondrial morphology and pyrimidine metabolism.“ Cell chemical biology25.3 (2018): 268-278.
12. Kauppila, Timo ES, Johanna HK Kauppila, and Nils-Göran Larsson. „Mammalian mitochondria and aging: an update.“ Cell metabolism 25.1 (2017): 57-71.
13. Gammage et al. “Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo” Nature medicine, 2017
www.nature.com/articles/s41591-018-0165-914. Emani, Sitaram M., et al. „Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury.“ The Journal of thoracic and cardiovascular surgery 154.1 (2017): 286-289.
www.jtcvs.org/article/S0022-5223 (17)30258-1/fulltext
15. McCully, James D., et al. „Mitochondrial transplantation: From animal models to clinical use in humans.“ Mitochondrion 34 (2017): 127-134.
www.sciencedirect.com/science/article/pii/S1567724917300053