Présentation
De quoi parle ce texte
Si une personne entend parler d'une «simulation de la réalité», alors il est probable qu'elle proposera diverses œuvres de science-fiction (telles que Matrix, The Dark City ou Zero Theorems) ou des jeux informatiques. Dans le cas de personnes dont la tête est obstruée par un diplôme d'ingénieur, des packages tels que
KOMPAS-3D AutoCAD, Solid Edge ou NX peuvent apparaître. Une personne qui écoute la science se souviendra probablement de toute
modélisation de divers engins spatiaux .
Mais il y a un niveau de Réalité de plus, qui s'avérera à juste titre oublié: celui auquel se déroule toute la chimie est le niveau des atomes et des molécules. Il peut également être simulé avec succès sur un ordinateur. Puisque la mécanique quantique est en charge de tout dans cette section de la réalité, de tels calculs sont souvent appelés chimie quantique. Et nous parlerons de sa connexion avec la Réalité étudiée par des méthodes expérimentales.
Ce texte portera sur les choses les plus élémentaires. Mais la pratique de lire des revues scientifiques et d'écouter divers rapports montre que cela doit être constamment rappelé.
Le texte est conçu pour les personnes qui comprennent et / ou s'intéressent à la façon dont vivent les atomes et les molécules.Tiré de xkcd.comBref historiqueIl se trouve que quelqu'un, malheureusement, a travaillé dans la science russe, m'a invité à donner une conférence à son cours spécial pour 2 personnes dans l'une des universités physiques bien connues de la Russie. Mais, par une étrange coïncidence, elle a été transférée à une conférence d'étudiants tenue en parallèle ... Là, elle n'a pas suscité beaucoup d'intérêt parmi les étudiants, et j'étais très, très désolée pour le matériel, j'ai donc décidé de spammer un peu Habr, essayant de transformer la conférence éducative en un article scientifique populaire.
Méthodes physiques pour étudier la vie des molécules
Nous savons par les cours de chimie et de physique de l'école que toutes les substances sont composées d'atomes, de molécules, d'ions ou de combinaisons de ceux-ci. Et nous semblons même savoir quel genre de vie ils vivent. Mais ces informations devraient avoir leurs propres sources fiables (méthodes de recherche), et elles le sont vraiment.
Il existe de nombreuses façons d'espionner la vie des atomes. Ceux qui le souhaitent, par exemple, peuvent se familiariser avec certains d'entre eux plus en détail dans les manuels classiques
- Pentin Yu.A., Vilkov L.V. Méthodes de recherche physique en chimie. - M .: Mir, 2006,
- Drago R. Méthodes physiques en chimie. - M .: Mir, 1981.
Mais, grosso modo et assez facilement, 3 groupes principaux de méthodes se distinguent:
- méthodes spectroscopiques
- méthodes de diffraction
- diverses méthodes de microscopie (peu importe, translucide ou à balayage, pour nous ce n'est pas essentiel maintenant).
On ne parlera pas de ce dernier, mais ses outils ne sont pas moins importants que les deux premiers.
Pourquoi on ne parlera pas de microscopie(Je n'ai pas du tout peur du mot en microscopie)
Méthodes spectroscopiques pour l'étude de la matière
Ce puissant groupe de méthodes nous offre de très, très nombreuses choses: de la recherche et de la détermination de molécules dans le milieu interstellaire et sur d'autres planètes au contrôle banal des explosifs à l'aéroport.
Le principe général des méthodes spectrales
Lorsque l'on parle de spectroscopie, se réfère généralement au principe général de fonctionnement suivant.
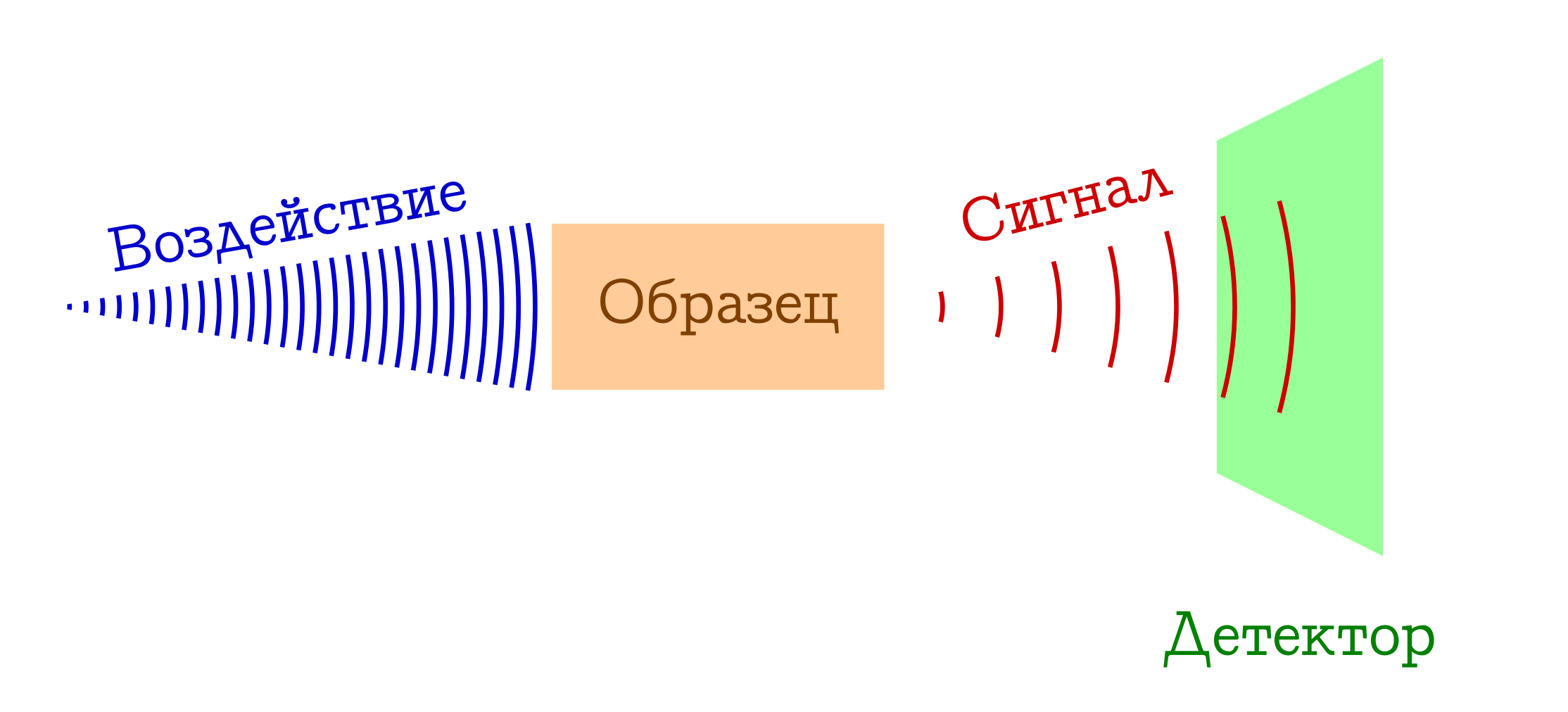
Le schéma général des méthodes spectrales pour l'étude des substances
- Nous avons quelque chose avec lequel nous (par exemple, une ampoule / laser / lumière du soleil) agissons sur l'échantillon qui nous intéresse. Le plus souvent, il s'agit d'une étude électromagnétique, mais il pourrait bien s'agir d'électrons (par exemple, en spectroscopie de masse avec ionisation par impact d'électrons) ou d'un cocktail de tout ce qui est possible et impossible à partir du plasma (par exemple, en spectroscopie de flammes , si apprécié des écoliers et des étudiants juniors de la faculté de chimie). D'une manière ou d'une autre, quelque chose doit fonctionner sur notre échantillon.
- Lorsqu'il est exposé à un échantillon, quelque chose se produit qui change son état. Cela peut être une transition vers une sorte de niveau excité (dans n'importe quelle spectrophotométrie ou spectroscopie Raman), ou même l'effondrement du système moléculaire (comme dans les spectres de masse ou la spectroscopie photoélectronique ). Mais en quelque sorte, le schéma à un moment donné devrait être différent.
- ???
- PROFIT !!! Nous enregistrons un certain signal (émis ou absorbé) avec ce changement dans l'échantillon au niveau moléculaire. Cela peut être des photons perdus dépensés pour changer l'échantillon (alors nous avons la spectroscopie d'absorption), ou vice versa, des photons en excès émis après excitation préalable de la substance (spectroscopie d'émission), un changement de la longueur d'onde des photons initiaux en raison de l'interaction avec la substance (spectroscopie Raman, plus connu à l'étranger sous le nom de
Ramenovskaya Ramanova ), ou stupidement des fragments des molécules originales (comme dans les spectres de masse ou la spectroscopie photoélectronique ). Il existe de nombreuses options - l'essence est la même: il y a un signal!
Comme exemple de telles méthodes, vous pouvez citer un tas de lettres différentes: RMN, ESR, MW, THz, IR, UV / Vis, XRF, MS, PES, EXAFS, XANES, etc. etc.
Tous (ou plusieurs d'entre eux) sont familiers (ou devraient être familiers) à chaque chimiste. Toutes ces méthodes sont l'arsenal standard (loin d'être incomplet) d'un chercheur qui se respecte et qui s'occupe des substances.
Gammes spectrales et leur relation avec la vie des molécules
 Tiré de xkcd.com
Tiré de xkcd.comÉtant donné que dans la très grande majorité des cas, la spectroscopie est toujours liée au rayonnement électromagnétique, il est logique de relier les plages du spectre électromagnétique à divers aspects de la vie atomique et moléculaire. Après tout, la fréquence des ondes électromagnétiques utilisées en spectroscopie est une sorte d '«horloge» qui vous permet de détecter la durée de tel ou tel processus dans les systèmes moléculaires. Ainsi, en changeant cette fréquence, vous pouvez étudier (et même agir) sur différents processus moléculaires.
Alors.
- Du point de vue chimique, rien d'intéressant ne se produit dans la gamme de très grandes longueurs d'onde, vous ne pouvez donc pas vous en souvenir.
- Avec la fréquence des ondes radio et micro-ondes (et même des rayons infrarouges à ondes longues, IR = IR), différentes molécules tournent dans la phase gazeuse: grandes et lourdes - dans la région des ondes radio (fréquences plus basses), et petites et légères - en IR (fréquences plus élevées).
- Cependant, dans l'IR, diverses vibrations moléculaires se produisent (principalement): tous les mouvements conformationnels et autres mouvements non évidents à l'intérieur des molécules se trouvent dans l'IR à longue longueur d'onde, et les vibrations d'étirement (étirement - raccourcissement des longueurs des liaisons chimiques) se produisent dans la courte longueur d'onde (jusqu'à 4000 cm -1 ).
- Eh bien, vient alors la place du spectre, où vivent diverses transitions électroniques (jusqu'à la région des γ-quanta). Aux basses fréquences (visible, UV = UV et rayons X mous), vivent principalement les transitions associées aux électrons de valence.
Pourquoi voyons-nous?Soit dit en passant, c'est précisément à cause des transitions électroniques que nous pouvons voir: dans nos yeux (dans les cônes), il y a des structures qui ont une
rétine dans leur composition. Lorsqu'un photon visible est absorbé par cette molécule, une double liaison s'y rompt, ce qui conduit à une isomérisation cis-trans. Et c'est ce changement que nous percevons comme le signal primaire, qui est ensuite transmis à notre cerveau.
Mais avec l'augmentation de l'énergie des photons (c'est-à-dire avec une fréquence croissante, comme nous le rappelons la formule de Planck E = h n u ), nous arrivons à des couches de plus en plus profondes de la structure électronique, jusqu'à ce que nous nous reposions dans la gamme de rayons X jusqu'aux derniers coquilles 1s ( ou, comme on les appelle rayons X, K ).
Ainsi, en choisissant la bonne longueur d'onde du rayonnement électromagnétique, nous pouvons examiner plus en détail un processus particulier dans les molécules.
Méthodes de diffraction des substances
Parlons maintenant un peu de diffraction. Le diagramme schématique de ces expériences est également simple.
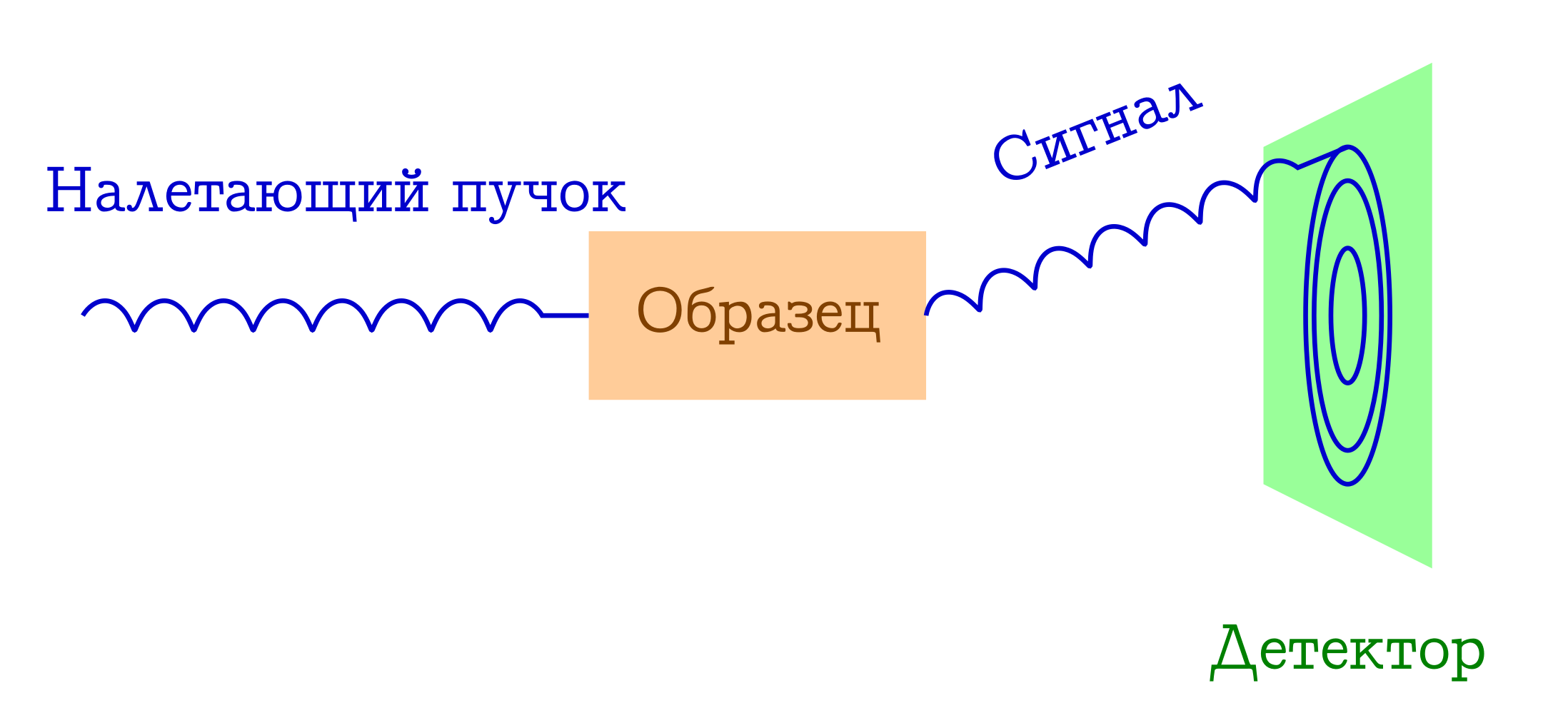
Schéma général des méthodes de diffraction pour l'étude des substances
- Un faisceau de quelques particules vole sur l'échantillon. Le plus souvent, ce sont soit des photons de rayons X, soit des électrons, soit des neutrons.
- Ces particules par différents mécanismes se dispersent élastiquement sur les atomes de l'échantillon qui nous intéresse (c'est-à-dire que sans changer la longueur d'onde et la phase de l'onde, elles changent simplement la direction de leur vol). Il ne se passe rien à l'échantillon de ces particules incidentes: il n'a tout simplement pas le temps de réagir.
- Les distances interatomiques servent de réseau de diffraction pour le faisceau incident, par conséquent, nous verrons une belle image de diffraction sur le détecteur.
Du dernier paragraphe, la condition pour la longueur d'onde des particules incidentes (λ) se pose: elle doit être du même ordre ou inférieure à l'ordre caractéristique des distances interatomiques, donc λ typique pour ces méthodes est de 1 à 0,01 Å.
Les principaux types d'erreurs lors de la comparaison des expériences et des calculs théoriques
En conséquence, nous avons une image très intéressante: en spectroscopie et en diffraction, nous observons une sorte de signal gauche, qui indique en quelque sorte
indirectement ce qui se passe réellement dans le système moléculaire.
L'analogie avec la grotte platonicienneCette peinture rappelle étrangement le
mythe de la grotte de Platon . Nous avons un certain monde réel de molécules. Mais nous n'en voyons que des ombres sur la paroi de la grotte (détecteur), qui sont un affichage incomplet de toutes les choses intéressantes qui se produisent à ce niveau de réalité.
Mais, heureusement, parfois nous pouvons théoriquement calculer le signal qui nous intéresse (comme, par exemple, en spectroscopie micro-ondes, IR ou UV / Vis), et parfois nous pouvons extraire du signal observé les quantités d'intérêt qui sont disponibles pour le calcul chimique quantique (par exemple, distance entre les atomes dans une molécule, moment dipolaire, etc.). Et ici, nous avons une chance que l'expérience numérique et l'expérience réelle puissent s'unir dans l'étape passionnée de la comparaison les uns avec les autres ... et ici 4 types d'erreurs peuvent se produire en standard.
Attention! Le terme «erreur» ne signifie pas ici que le résultat de la comparaison est manifestement erroné. C'est juste que le terrain de comparaison devient très fragile et marécageux, et une étape bâclée peut facilement gâcher tout le travail.
- Différentes conditions de l'expérience et / ou du calcul (état d'agrégation, température, pression, etc.). Nous pouvons soudainement commencer à comparer différents systèmes entre eux, pour une raison quelconque, en les considérant comme les mêmes. Par exemple, il est évident que l'ajout d'une ou cinq cuillères à café de sucre à une tasse de thé conduira au même système physique appelé «thé au sucre», mais les propriétés de ce système seront très différentes. Et il peut être facilement mesuré. Par exemple, avec un thermomètre (mesure de la température du thé immédiatement après la dissolution du sucre) ou avec la langue (l'une des méthodes d'analyse dites organoleptiques). Ainsi, en comparant les systèmes résultants entre eux (que ce soit une vraie tasse de sucre avec du thé ou son modèle informatique), nous ne devons pas oublier que les similitudes ont leurs limites, et que si nous réduisons la marge d'erreur pour la «similitude», nous finirons par trouver les différences.
- Signification physique et / ou mathématique différente des paramètres (le paramètre de la signification physique au sens habituel peut même ne pas exister). Ici aussi, tout est simple: si nous comparons 2 quantités avec un nom similaire, cela ne signifie pas que les quantités ont la même signification physique. Par exemple, la cote de l'adjoint parmi l'ensemble de la population de la ville contre évaluation uniquement parmi les mamies. Tant cette cote que cette note (quelle qu'elle soit), ces chiffres (ou quoi que ce soit) peuvent même fortement corréler les uns avec les autres, mais la signification de ces paramètres est toujours différente, et cette différence peut être détectée.
- Erreurs "aléatoires" . Cela inclut des erreurs systématiques dont l'expérimentateur / théoricien du simulateur n'a pas connaissance, ou des erreurs vraiment aléatoires dans l'expérience / le calcul qui ne peuvent pas être contrôlées et / ou prédites. En principe, de telles choses peuvent elles-mêmes devenir l'objet d'une enquête sur divers effets systématiques intéressants.
ou simplement une estimation du rapport S / N («signal sur bruit») le plus utile. - Et la dernière erreur standard est la croissance des mains de l'expérimentateur / calculateur à partir de l'os pelvien , c'est-à-dire des erreurs humaines ordinaires. Il n'est pas nécessaire d'enquêter sur quoi que ce soit, il suffit de revérifier le travail ou de répéter l'expérience pour trouver et éliminer le montant correspondant.
Rien de plus concret ne peut être dit sur les deux derniers types d'erreurs, mais beaucoup peut être dit sur les deux premiers, mais si vous prenez une méthode de recherche spécifique. Par conséquent, nous nous concentrerons sur eux. Dans ce cas, l'accent sera principalement mis sur les différences structurelles des molécules.
Erreur # 1. Différences de propriétés moléculaires dans différentes conditions
NaCl: quand aucune erreur
Pour une raison quelconque, personne ne pense que le monocristal de chlorure de sodium (NaCl), qui est une énorme molécule d'ions Na
+ et Cl
- , et la molécule diatomique NaCl, obtenue par évaporation de ce cristal à des températures folles, ont un seul et même disons la structure.
Et même si nous supposons qu'au moins les distances entre le chlore et le sodium (
r NaCl ) sont les mêmes ici et là, l'expérience nous mettra en place:
Où commettons-nous une erreurEn fait, avec une telle comparaison, nous permettons la possibilité de l'erreur # 2, mais tout va bien ici, si nous évaluons les erreurs d'une telle comparaison, elles seront de l'ordre de 0,01 Å, ce qui est nettement inférieur à la différence des paramètres comparés. C'est-à-dire ce n'est pas une erreur, mais un effet réel.
Comment obtenir vous-même la distance entre le cation sodium et l'anion chlore dans un cristal de selL'obtention de la distance entre les atomes dans une molécule de NaCl diatomique à partir de données expérimentales n'est pas non plus une procédure aussi compliquée. Mais le problème est seulement qu'une telle expérience est une chose compliquée. Par conséquent, il est plus facile d'utiliser une base de données où les distances requises sont déjà données.
Mais pour obtenir la distance entre les atomes dans le cristal, seule la densité du sel cristallin du tableau ρ = 2.165 g / cm
3 suffit, ce qui peut
être facilement obtenu sur Wikipedia et mesuré soi-même à la maison.
Pour calculer la distance dont nous avons besoin:
- la densité du cristal de NaCl (is),
- connaissance de la localisation des ions de ce cristal.
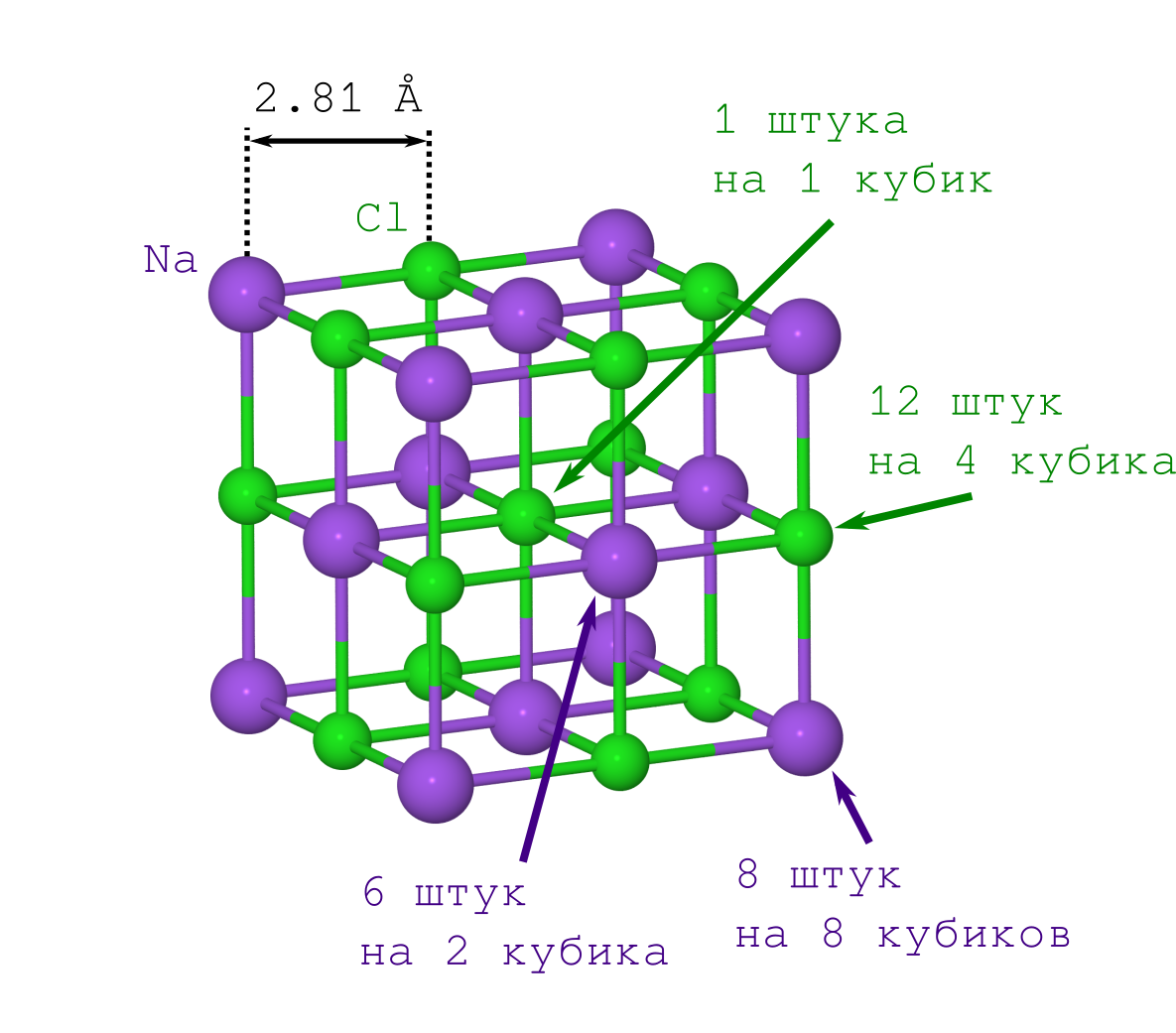
Si vous l'aviez fait pour la première fois (disons, au début du 20e siècle), vous auriez à vous tourmenter avec le deuxième point. Mais cela est déjà connu des gens modernes: le réseau NaCl a la forme d'un cube dans lequel les ions Na
+ et Cl
- alternent (voir l'image ci-dessus). En multipliant le fragment indiqué du cristal («copier-coller» la pièce spécifiée et en l'ajustant face à face à l'itération précédente), nous obtenons un cristal de NaCl de toute taille souhaitée et de toute forme (minecraft) souhaitée.
La densité de ce cube devrait donc être la même que celle du cristal entier. Étant donné que la densité est
rho= fracmV (c'est-à-dire masse par volume), il s'avère que connaissant la masse et l'expression géométrique du volume, nous pouvons calculer la distance entre les atomes.
Le volume du cube est évident: la longueur de la nervure est le double de la distance Na - Cl (
L=2r mathrmNaCl ), ce qui signifie que le volume souhaité est
V=L3=8r mathrmNaCl3 .
La masse n'est pas si simple. La plupart de nos atomes se trouvent sur les sommets, les bords et les faces du cube, ce qui signifie qu'ils appartiennent simultanément à plusieurs de ces cubes. Cela doit être pris en compte dans les calculs.
Commençons par les ions Na
+ . Nous n'en avons que 2 types (voir le modèle de réseau cristallin):
- ceux qui sont aux sommets du cube (il y en a autant que les sommets du cube, c'est-à-dire 8, et ils sont simultanément en 8 cubes, vous devrez donc diviser ce nombre par 8),
- ceux qui se trouvent sur les visages (il y en a 6, et ils appartiennent simultanément à 2 cubes).
En conséquence, nous obtenons que notre cube contient

ion sodium.
Maintenant sur Cl
- . Il existe également seulement 2 types d'entre eux (voir le modèle de réseau cristallin):
- celles qui se trouvent sur les bords du cube (il y en a 12, et elles sont détenues conjointement par 4 cubes),
- ce Cl - qu'au centre du cube, il est un et n'appartient qu'à notre cube.
Par conséquent, notre cube contient

ion chlore.
La composition du cristal correspond évidemment à la formule chimique du NaCl, mais la masse de notre cube est égale (n'oubliez pas que les masses d'atomes du tableau périodique sont données en
unités de masse atomique ):
m=4 cdot( underbraceM mathrmNa23 textam+ underbraceM mathrmCl35.5 textamu)=234 textamu=234 cdot1.66 cdot10−24 textg=3.88 cdot10−22 textg\.
Maintenant de la relation
rho= fracmV nous pouvons faire une équation pour la longueur
r mathrmNaCl :
r mathrmNaCl3= left( fracm8 rho right) ,
qui se résout facilement:
r mathrmNaCl= left( fracm8 rho right)1/3= left( frac3.88 cdot10−22 [ textg]8 cdot2.17 [ textg/ textcm3] right)1/3=2,82 cdot10−8 [ textcm]=2,82 [ textÅ] .
D'après les données de la cristallographie aux rayons X 2,81 Å (par exemple, d'
Abrahams, SC; Bernstein, JL Précision d'un diffractomètre automatique. Mesure des facteurs de structure du chlorure de sodium // Acta Crystallographica (1965) 18, 926-932 ), nous n'avons manqué que 0,01 Å, ce qui est assez cool.
Quelqu'un pourrait penser que la différence de 0,45 Å est insignifiante, mais c'est presque le rayon de Bohr (0,52 Å), qui est égal à la distance la plus probable de l'électron, et selon les normes atomiques, la différence est énorme.
Pourquoi NaCl sous la forme d'une molécule atomique 2 diffère d'un cristalIci, tout est très simple. Le réseau cristallin infini crée la possibilité d'un «saut» irréversible pour 3s
1 électron de sodium par atome de chlore, car la différence de charge résultante est compensée par l'interaction avec les voisins.
3s
1 ( ), ,
«» :
Na:Cl↔Na+Cl−
() (), .
,
±1 , , .
NaCl (2.36 Å),
d=q⋅rNaCl=9.0 [] où
q≥0 (
+q ,
−q )
, « » 0.21, ..
d=0.21⋅9.0=1.9 [qe⋅Å] , :
q=drNaCl=1.92.36=0.8 . «» 0.2 NaCl NaCl .
Ferrocène
Il vaut la peine de passer des cristaux ioniques à des cristaux moléculaires dans lesquels les molécules sont densément emballées, donc soudainement il devient possible de comparer, et sans aucune réserve.Mais la différence ne doit pas être oubliée. Et il existe même un exemple classique à ce sujet: une molécule de ferrocène .Il s'agit de la connexion sandwich la plus simple. Dans celui-ci, un atome de fer neutre (comme une côtelette) est pris en sandwich entre deux anneaux aromatiques à cinq membres (brioches).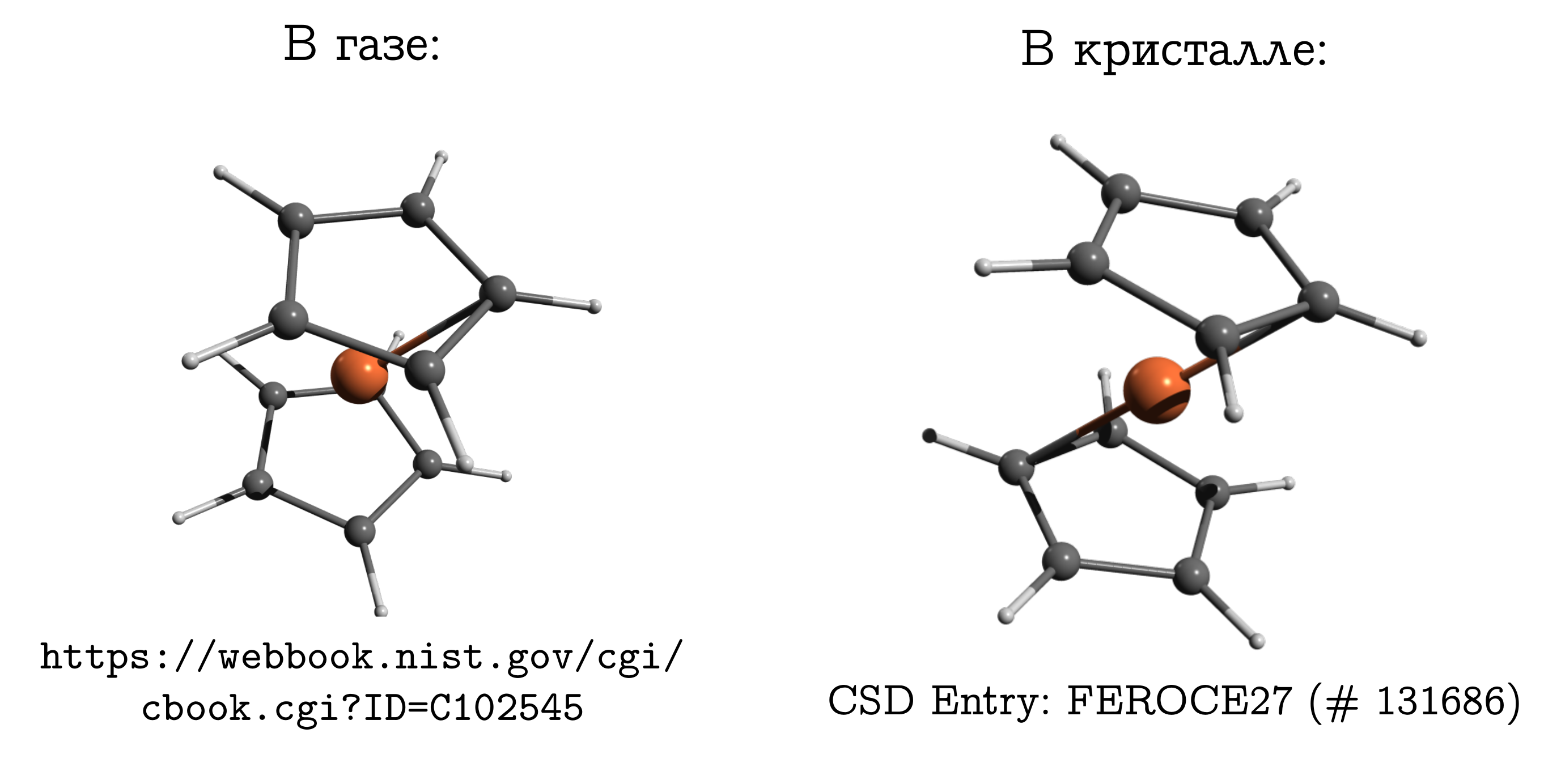 Cette molécule peut être évaporée assez facilement et découvrez que la structure dite la plus stable en phase gazeuse est obstruction obstruée. Dans celui-ci, les carbones et les hydrogènes des anneaux supérieur et inférieur sont opposés (voir l'image ci-dessus), car dans ce cas, les interactions de dispersion sont les plus fortes entre ces morceaux de la molécule, et la dispersion est toujours bénéfique.Si nous prenons un cristal de ferrocène, il s'avère que les molécules y ont une conformation stable différente (qui est appelée inhibée pour les hydrocarbures), dans laquelle l'hydrogène et le carbone d'un cycle sont au-dessus / sous la liaison C-C de l'autre. Il existe des interactions de dispersion entre les molécules et une structure similaire, apparemment gênante pour une structure moléculaire, découle du fait qu'il est plus facile pour les molécules de s'emboîter uniquement sous une forme inconfortable, et cet inconvénient personnel est compensé par une interaction les uns avec les autres.
Cette molécule peut être évaporée assez facilement et découvrez que la structure dite la plus stable en phase gazeuse est obstruction obstruée. Dans celui-ci, les carbones et les hydrogènes des anneaux supérieur et inférieur sont opposés (voir l'image ci-dessus), car dans ce cas, les interactions de dispersion sont les plus fortes entre ces morceaux de la molécule, et la dispersion est toujours bénéfique.Si nous prenons un cristal de ferrocène, il s'avère que les molécules y ont une conformation stable différente (qui est appelée inhibée pour les hydrocarbures), dans laquelle l'hydrogène et le carbone d'un cycle sont au-dessus / sous la liaison C-C de l'autre. Il existe des interactions de dispersion entre les molécules et une structure similaire, apparemment gênante pour une structure moléculaire, découle du fait qu'il est plus facile pour les molécules de s'emboîter uniquement sous une forme inconfortable, et cet inconvénient personnel est compensé par une interaction les uns avec les autres.Pourquoi le ferrocène est si différent de l'éthaneUne personne familière avec la chimie doit généralement se maîtriser pour se souvenir de la structure du ferrocène dans un gaz. Après tout, il a des souvenirs d'éthane (C
2 H
6 ), dans lequel la conformation la plus stable est inhibée (lorsque les hydrogènes d'un morceau de CH
3 se trouvent «entre» les hydrogènes d'un autre CH
3 ), parce que dans cette position, la répulsion interatomique entre les enveloppes d'électrons des hydrogènes est minimisée.

Adapté de
www.chem.msu.su/rus/teaching/stereoEt ici, toute la différence est dans la distance. La forme standard du potentiel d'interactions de dispersion est le potentiel de Lennard-Jones (soit dit en passant, c'est un, pas deux hommes):
V mathrmLJ(r)= fracAr12− fracBr6
Dans ce document, le premier terme est tiré de la répulsion interatomique et le second de l'attraction interatomique résultant des fluctuations de la densité électronique. En général, ce potentiel ressemble à ceci:
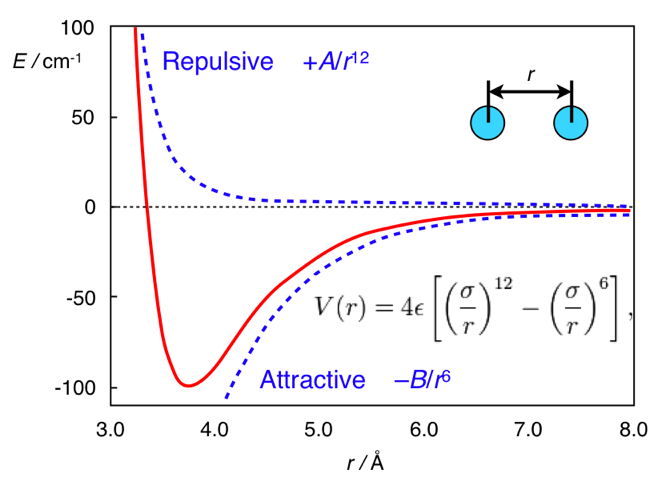
Potentiel de Lennard-Jones. Adapté de
chemistry.stackexchange.com/questions/34214/physical-significance-of-double-well-potential-in-quantum-bondingEt dans le cas de l'éthane, les atomes d'hydrogène sont trop proches les uns des autres, ils sont donc (par rapport à son minimum) sur le côté gauche de la courbe, et ils sont caractérisés par la répulsion. Dans le cas du ferrocène, entre les anneaux, il y a une couche de taille non maladive (un atome de fer), grâce à laquelle les anneaux sont suffisamment éloignés pour ne pas ressentir de répulsion interatomique. Et donc ils sont sur la partie droite (attrayante) du potentiel.
Histamine
Dans le cas du ferrocène, nous avons vu le soi-disant différences de conformation: la molécule est restée la même (c'est-à-dire qu'aucune liaison chimique n'a été rompue ou formée) et sa forme a légèrement changé.
Mais les différences peuvent être encore plus fortes, par exemple, si le soi-disant
transformations tautomères . La tautomérisation est une classe de réactions chimiques qui se produisent si facilement et rapidement qu'en conséquence, nous pouvons avoir simultanément plusieurs isomères d'une molécule, se passant facilement les uns dans les autres. Ces isomères sont appelés tautomères.
Un exemple standard de ceci: le tautomérisme céto-énol dans les cétones:

Le plus souvent, comme dans cet exemple, le tautomérisme est associé au saut d'un proton d'un endroit chaud à un autre. Et ces réactions sont liées à l'
effet tunnel , auquel l'hydrogène, le plus léger des atomes, est le plus sensible.
De telles transformations chimiques sont caractéristiques de nombreuses molécules biologiques, par exemple les
bases azotées qui composent l'ADN ou les
sucres .
Mais en passant d'un système à un autre, les constantes d'équilibre de ces réactions changent souvent, donc dans différentes phases nous pouvons observer différentes compositions tautomères.
Un exemple de ceci est la molécule d'histamine (voir figure ci-dessous).
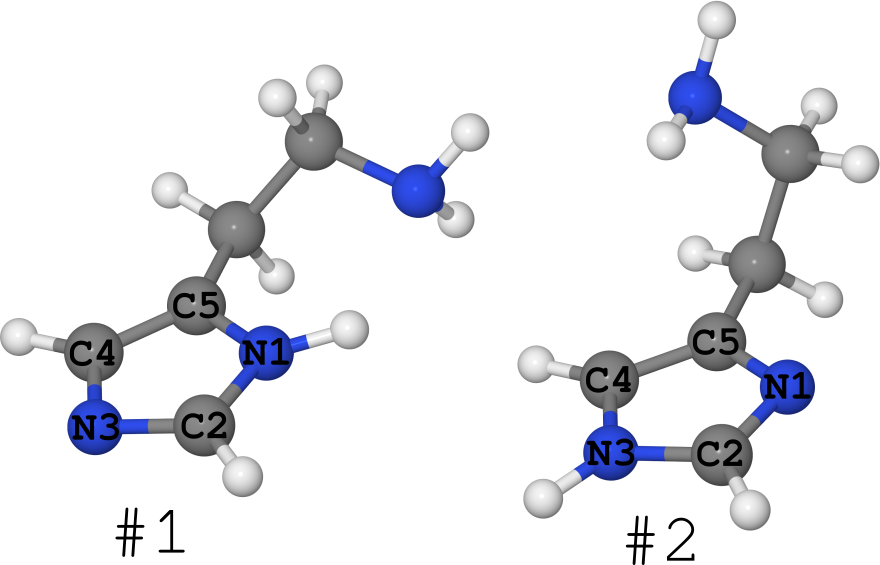
Il existe sous la forme de 2 tautomères (je suis généralement silencieux sur le nombre de conformers, il y en a beaucoup):
- # 1, où l'hydrogène repose sur l'azote N1,
- # 2, où l'hydrogène se trouve sur l'azote N3.
Il se trouve que pour cette molécule, ses structures en différentes phases sont connues.
- Dans le cristal, il est complètement "figé" sous forme de # 1. (voir l'article DOI: 10.1021 / ja00796a011 et la structure de la Cambridge Structures Bank sous le nom "HISTAN" et / ou numéro 1176642)
- Dans les solutions aqueuses, cette molécule existe sous les deux formes, et le tautomère # 2 est sensiblement plus grand ( DOI: 10.1021 / ja027103x ).
- Dans le gaz, l'histamine existe également sous la forme # 1 et sous la forme # 2 ( DOI: 10.1021 / ja980560m ).
C'est-à-dire différentes phases contiennent différents nombres de molécules différentes, ce qui signifie que ce sont des systèmes différents.
Conclusion d'erreur # 1

La principale conclusion à tirer des exemples ci-dessus est la suivante:
Lors de la comparaison de calculs dans une phase avec une expérience dans une autre, il faut être préparé aux différences systématiques.
Cela ne signifie pas qu'il n'est pas nécessaire de comparer: il faut comparer, mais il faut juste être plus critique vis-à-vis des différences et / ou des coïncidences trouvées, et évaluer si possible ces effets.
Erreur # 2. Paramètres moléculaires "Zoo".
La deuxième erreur est brièvement décrite comme suit: si les paramètres sont appelés de manière similaire, mais pas identique, ce sont des paramètres différents.
Pour comprendre quelle est la source de ce désaccord entre théorie et expérience, il faudra analyser plus en détail à la fois les méthodes expérimentales standard utilisées pour obtenir les paramètres moléculaires et les modèles qui calculent des quantités similaires uniquement à partir de la théorie.
Et ici, nous ne parlerons à nouveau que des structures.
Comment obtenir des structures moléculaires expérimentales
Afin de nous limiter en quelque sorte, nous ne parlerons que de méthodes pour étudier la structure de molécules uniques, c'est-à-dire sur la phase gazeuse.
Nous avons deux sources principales de telles informations:
- diffraction d'électrons gazeux,
- spectroscopie micro-ondes.
Nous nous attarderons sur chacune de ces méthodes plus en détail.
Diffraction d'électrons gazeux
La méthode est assez ancienne, elle remonte aux années 30 du XXe siècle, lorsque les scientifiques allemands Mark et Wirl ont réalisé les premières expériences sur la diffraction des électrons par le gaz.
Peu de gens le savent, mais cette méthode de recherche est impliquée dans l'obtention de trois prix Nobel de chimie.
3 nobles avec entrée électronique- Peter Debye en 1936 a reçu son prix avec le libellé:
"[pour ses travaux sur] la structure moléculaire à travers ses recherches sur les moments dipolaires et la diffraction des rayons X et des électrons dans les gaz "
C'est la seule mention explicite de la diffraction des électrons gazeux au mérite du lauréat, et non sans raison. L'équation de diffraction électronique de base pour l'intensité de diffusion moléculaire est nommée Debye.
en fait, l'équation de DebyeIij(s)=gij frac sin(srij)rij
Ici
Iij désigne l'intensité de diffusion des électrons (ou rayons X, ou d'autres particules) par une paire de
i- ème et j-ème atomes à distance
rij à part
s= frac2 pi lambda sin left( frac theta2 right) La coordonnée de diffusion est-elle associée à l'angle de diffusion
theta et longueur d'onde des particules
lambda et
g - la capacité de cette paire d'atomes à diffuser des particules diffractantes.
Et malgré le fait qu'on se souvienne de quelque chose de cette merveilleuse physique (le modèle des solutions ioniques , son modèle de calcul de la capacité thermique des cristaux ), mais pas de la diffraction électronique, il a reçu le principal prix scientifique (en particulier) pour cela.
- Linus Pauling en 1954. Oui, celui qui a reçu 2 prix Nobel personnels,
et même mis le monde entier sur la vitamine C , Great Pauling. Lorsqu'il travaillait à Kaltekh, en particulier, il était engagé dans la diffraction d'électrons gazeux (voir, par exemple, DOI: 10.1021 / ja01873a047 ). Et bien sûr, sa connaissance de la chimie structurale des molécules libres l'a aidé à créer la fameuse théorie de la liaison chimique (mais ne minimisons pas ici son vaste arrière-plan cristallographique). - Odd Hassel, lauréat 1969. Il a reçu son 1/2 prix Nobel pour la découverte de l'équilibre conformationnel. Il l'a fait sur la base d'une étude de diffraction d'électrons sur le cyclohexane. Cette molécule existe sous forme de deux conformations: une chaise (chaise) et une salle de bain (dans la tradition anglaise - un bateau, un bateau).

À partir d'ici: www.shapeways.com/product/N5FE298DS/cyclohexane-2-molecules-boat-and-chair-form
Ces options pour l'arrangement des atomes se transforment rapidement l'une en l'autre, mais à cette époque, elles ne le savaient pas et croyaient qu'une seule des structures devait être réalisée. Seul le signal de diffraction d'électrons ne voulait être décrit par aucune de ces structures, et seule une combinaison de signaux des deux conformations pouvait expliquer le schéma de diffraction observé (plus d'informations à ce sujet peuvent être trouvées dans le livre de I. Khargittai "Frank Science. Conversations with Famous Chemists").
Le schéma de la méthode elle-même est très simple (voir l'image ci-dessous).
La chose se passe dans le vide.
- Des électrons rapides sont continuellement éliminés de la cathode, qui sont accélérés dans le champ anodique à des énergies de 40 à 60 keV.
- Les électrons suffisamment dispersés (mais rapides) sont focalisés par une lentille magnétique, après quoi ils se transforment en un faisceau étroit.
- Une chambre avec une substance est installée perpendiculairement à la poutre. L'échantillon est chauffé à ébullition et la vapeur résultante entre en contact avec le faisceau d'électrons.
- Les électrons sont correctement dispersés par les molécules et s'envolent tranquillement plus loin, où ils tombent sur le film.
- Habituellement, devant le film mettre le soi-disant. périphérique secteur. Il s'agit d'un écran tournant très rapidement d'une forme inhabituelle. Le fait est que l'électron a la probabilité de s'écarter de sa direction d'origine (par un grand angle de diffusion t h e t a ), tombe très rapidement. Par conséquent, afin de lisser cette diminution d'intensité, le secteur éclipse uniformément la partie centrale du film, laissant la partie éloignée ouverte. Le résultat est une image plus uniformément éclairée.
- Le piège à faisceau capte les électrons qui ne sont pas du tout diffusés (et il y en a beaucoup).
- Eh bien, pour que les molécules ne volent pas partout dans l'appareil, le salissant, elles sont congelées dans un piège froid refroidi par de l'azote liquide.
Le résultat est le même schéma de diffraction d'anneaux concentriques décrit par l'équation de Debye (c'est un signal). Divers paramètres moléculaires peuvent alors en être extraits directement.
Où puis-je trouver des laboratoires d'électrons à gaz?Il n'en reste plus tant.
Mais en Russie, il y en a deux: Moscou (au Département de chimie de l'Université d'État de Moscou) et à l'Université chimique et technologique Ivanovo.
Spectroscopie hyperfréquence
Cette méthode d'étude des molécules est plus connue, je vais donc en parler un peu plus brièvement, en prenant comme exemple la modification la plus moderne: un spectromètre à transformée de Fourier (comme en russe, en bref spectroscopie hyperfréquence transformée de Fourier).
La conception ici est déjà plus compliquée, car elle nécessite un tas d'électronique différente (amplificateurs, modulateurs de fréquence, etc.). Nous allons omettre tout cela et ne parler que de ce qui se passe à l'intérieur de la chambre à vide.
- En face de l'autre, il y a deux antennes à corne (comme celle qui a ouvert l' étude relique ). L'un d'eux sert d'émetteur et le second est un récepteur.
- Perpendiculaire à ces antennes se trouve une valve qui lance l'échantillon. Le plus souvent, il est lancé sous forme de vapeur avec un certain gaz vecteur (généralement des gaz inertes) en mode d'expansion adiabatique. Dans de telles conditions, les molécules refroidissent rapidement à des températures proches de 0 K, ce qui simplifie considérablement le spectre, le rendant plus sensible à l'interprétation.
- Lorsque les molécules remplissent toute la chambre, l'antenne d'émission les irradie avec un signal modulé en fréquence linéaire. Dans la représentation des fréquences, cela correspond à la somme de toutes les fréquences dans une certaine plage.
- Certaines molécules absorbent ce rayonnement à différentes fréquences transmises et entrent dans un état excité. Mais, après un certain temps, ils retombent, commençant à rayonner ce qu'ils ont capté pendant l'impulsion de l'antenne d'émission. Ce décrochage ressemble à un signal d'oscillation décroissant ( décroissance d'induction libre ). La deuxième antenne l'enregistre également. Puis, après la transformée de Fourier d'enregistrement de ce signal dans le temps, le spectre de fréquence habituel est obtenu.
Contrairement à la diffraction des électrons, qui n'était pas important quel type de molécules à considérer, dans une spectroscopie micro-onde, une molécule doit avoir un moment dipolaire constant (dans de rares cas, un moment dipolaire magnétique convient également, cela est typique pour les radicaux, comme une molécule d'O
2 ). Le signal ici est «intensité d'émission vs. fréquence. " Les constantes rotationnelles sont extraites de ces spectres à travers certains modèles, dont la structure moléculaire est ensuite extraite.
Bienvenue au Zoo des paramètres moléculaires!
Il est maintenant temps de regarder quels paramètres géométriques nous pouvons obtenir de diverses expériences. En fait, chacun des types de quantités indique quel type de modèle a été utilisé pour ajuster le signal expérimental (le plus souvent par la méthode des moindres carrés). La plupart de ces paramètres peuvent être trouvés dans la revue Kuchitsu K., Cyvin SJ // Dans: Molecular Structure and Vibrations / Cyvin SJ (Ed.) - Amsterdam: Elsevier, 1972.- Ch.12. - P.183-211.
Reprenons l'électronique.
- rg= langler rangleT la structure. Il s'agit simplement d'un ensemble de valeurs moyennes des distances interatomiques à une température donnée.
- ra= langler−1 rangle−1T cette valeur est similaire rg , mais c'est un peu plus naturel pour décrire le diagramme de diffraction.
- r alpha=rh,0=? . Cette valeur n'a pas de signification physique claire et est entièrement liée au modèle interprétatif. En fait, c'est ce qui est observé en cristallographie.
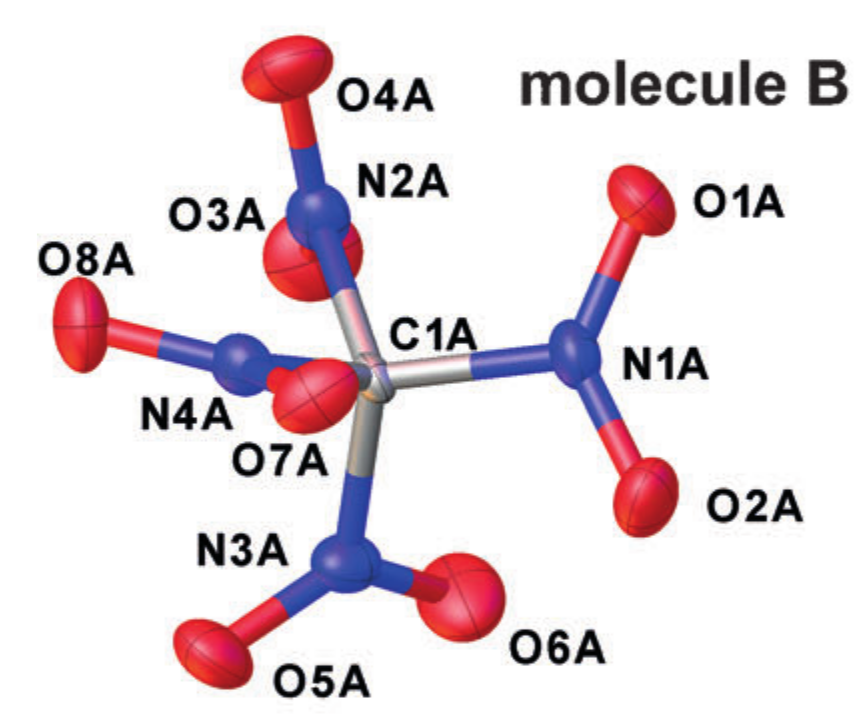
Exemple cristallographique r alpha structures (tétranitrométhane dans le cristal). Adapté de DOI: 10.1002 / anie.201704396
Chaque atome est approximé par un ellipsoïde décrivant son mouvement vibratoire, et les distances entre les centres des ellipses résultantes sont prises comme des distances interatomiques. Mais, une telle simplification de la nature du mouvement des atomes correspond à l'introduction de l'approximation de l'oscillateur harmonique pour les oscillations, et elle ne fonctionne pas toujours bien.
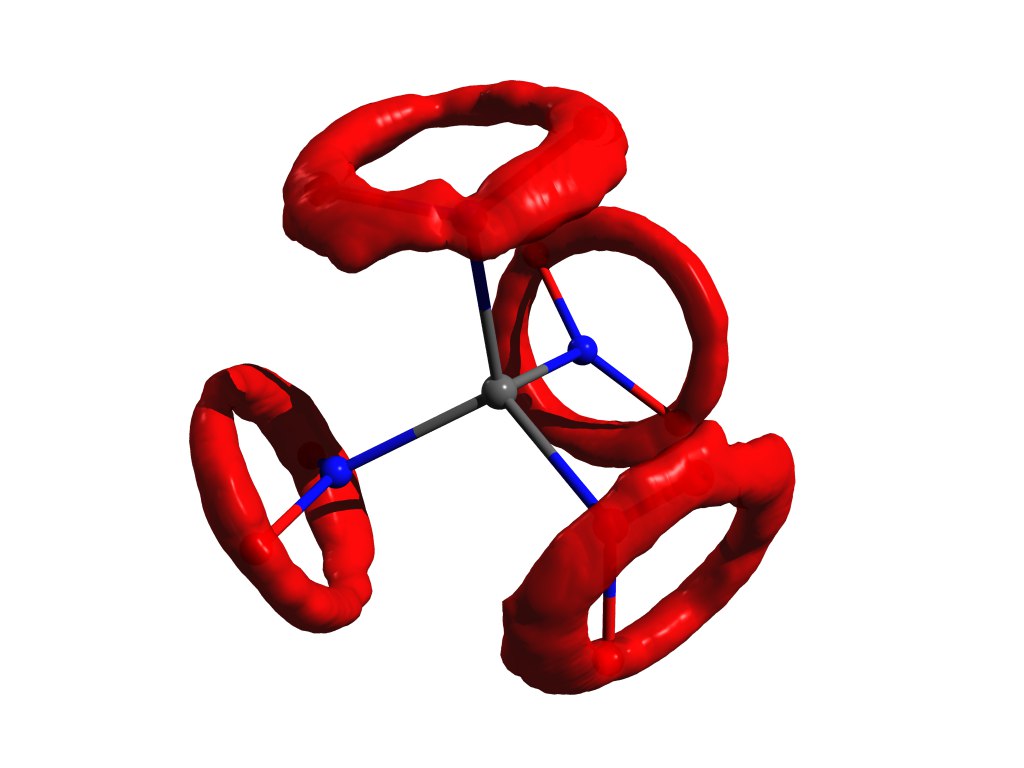
Un exemple de la distribution des atomes lorsque l'approximation de l'oscillateur harmonique ne fonctionne pas. Le même tétranitrométhane, mais dans le gaz.
- rh1=??? . Ce poisson-baleine de
judo miracle HEX n'est pas décrit en un mot, il a une relation très faible avec la réalité, mais il a de merveilleuses propriétés: il doit être géométriquement cohérent (voir ci-dessous) et il peut être facilement calculé. Pour cette raison, elle a gagné sa grande popularité dans la communauté électronique.
En spectroscopie micro-ondes, il existe un peu moins de variantes structurales.
- Le plus compréhensible physiquement est rn= langlen| chapeaur|n rangle la structure. En fait, c'est la géométrie moyenne sur un certain état vibratoire de la molécule ( |n rangle ) Puisqu'ils travaillent le plus souvent avec des molécules froides, ils observent généralement une structure spécifique de cette classe: r0 , c'est-à-dire la géométrie de la molécule à l'état vibratoire fondamental, lorsque les atomes ne produisent aucune vibration autour de leur position la plus avantageuse.
- Le plus populaire est rs la structure. L'indice «s» signifie «substitution». Ils l'obtiennent de cette façon: ils croient qu'il y a des coordonnées d'atomes qui sont fixes dans l'espace, puis ils font une substitution isotopique mono-atomique d'un atome dans la molécule, et ils déterminent la position de cet atome en changeant les constantes de rotation. Le principal avantage de cette technologie est la simplicité. Moins: vous avez seulement besoin d'une monosubstitution + toutes les positions des atomes ne peuvent pas être établies comme ça + la signification physique d'un tel modèle n'est pas très claire.
- Développement logique rs les structures sont rm -structures obtenues par ajustement par moindres carrés pondérés en fonction de la masse. Ils ont également besoin de molécules substituées isotopiquement, mais aucune d'entre elles ne convient déjà.
Et c'est loin de tous les types de structures possibles ...
Mais le
grand simulateur L' utilisateur d'un package standard de chimie quantique (tel que le programme
Gaussian Evil Corporation ) lors de l'utilisation d'un sort magique comme «Opt» obtient ce qu'on appelle la «géométrie d'équilibre», ou
re la structure. Il s'agit de la configuration la plus optimale des noyaux, minimisant l'énergie électronique du système. Et de telles structures peuvent également être retirées de la diffraction électronique et de la spectroscopie rotationnelle, mais uniquement pour des molécules très petites et symétriques, et en combinaison avec d'autres méthodes de recherche. Jusqu'à présent, cela ne fonctionne pas.
Et donc la question se pose: est-il correct de comparer
re structure avec une partie de l'expérimentation, en regardant uniquement les erreurs expérimentales?
La réponse ici est simple:
non , il faut faire une erreur supplémentaire sur d'éventuelles différences systématiques. Et un exemple très frappant peut être donné: l'effet Bastiansen-Morino (voir les articles
DOI: 10.1107 / S0365110060002557 et
DOI: 10.1107 / S0365110060002545 ).
Supposons que nous ayons une molécule de type CX
2 (c'est-à-dire CO
2 , CS
2 , etc.). Comme nous devons le savoir au cours de la chimie scolaire, ces molécules ont une structure linéaire (les atomes de carbone et deux chalcogènes X reposent sur une seule ligne droite).
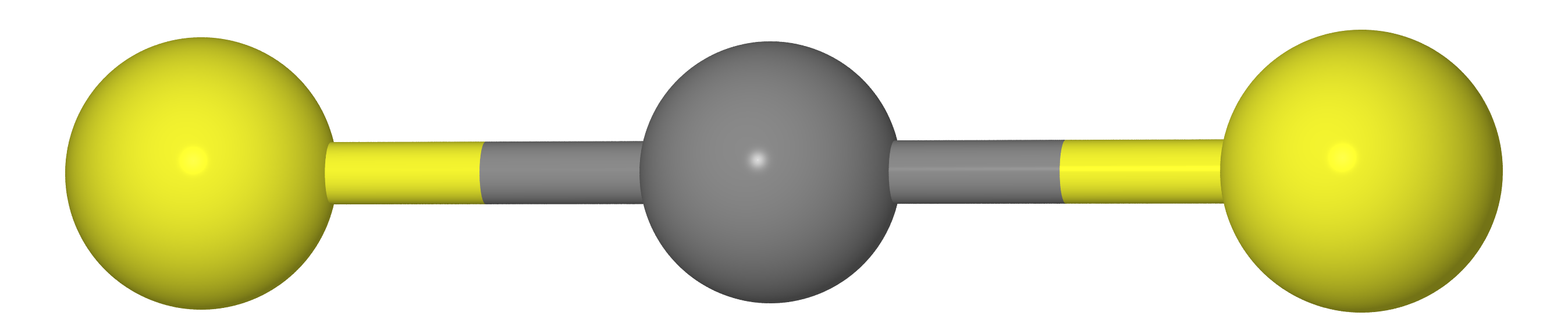
Cela signifie que la distance entre les atomes X doit être égale à deux fois la longueur de la liaison C - X (c.-à-d.
re( mathrmXX)=2re( mathrmCX) )

Quoi qu'il en soit, si nous mesurons les distances entre les atomes C et X (
rg( mathrmCX) ) et XX (
rg( mathrmXX) ) par diffraction d'électrons gazeux, on obtient que
rg( mathrmXX)<2rg( mathrmCX) , c'est-à-dire la molécule se révèle incurvée. La raison réside dans le fait que la molécule fait ce qu'on appelle
les vibrations des ciseaux , en raison desquelles les atomes X sont beaucoup plus proches les uns des autres que dans l'emplacement le plus favorable (voir la figure ci-dessous).

D'où vient l'effet Bastansen-Morino. Image tirée de l'article
DOI: 10.1039 / C6CP05849C .
Par conséquent, si nous égalisons la température moyenne
rg -structure à l'équilibre (
re ), nous ferions la mauvaise conclusion que les molécules de dioxyde de carbone et de disulfure de carbone sont courbes.
C'est pourquoi lorsque vous comparez différents types de paramètres géométriques, vous devez toujours être très prudent. Cela s'applique à la fois à une comparaison des données expérimentales entre elles et à une comparaison de l'expérience et de la théorie.
Molécules Molécules de modèle standard
Imaginons maintenant que nous voulions de tout cœur simuler le résultat d'une expérience sur la base de notre modèle théorique afin de comparer la simulation avec la réalité en combat loyal.
Et ici, il faut aussi être prudent, car différents modèles de molécules ont également leurs limites d'applicabilité. Examinons cela en utilisant l'exemple du modèle de molécule standard.
Vous devez d'abord comprendre ce qu'est le modèle standard de molécule. Les physiciens BAK ont leur propre
modèle standard , les astronomes ont
leur propre modèle et les physiciens ont leur propre conception de base, à partir de laquelle ils dansent plus tard. Mais contrairement aux modèles physiques, ce que nous considérons est un ensemble d'approximations qui permettent à l'utilisateur d'obtenir le résultat de manière relativement automatique et rapide.
Pour les utilisateurs gaussiensNous rappelons maintenant ce qui est à la base des sorts magiques gaussiens «Opt» et «Freq».
Le schéma général des approximations introduites ressemble à ceci:
Au bas de la qualité se trouve notre modèle standard. Passez brièvement en revue toutes les étapes de sa réception.
Le modèle résultant est appelé RR-HO (@BO). Nous ne toucherons pas à l'approximation de Born-Oppenheimer (BO), mais nous devrons parler du rotateur dur et de l'oscillateur harmonique dans le cadre de la chimie structurale ...Et le principal problème avec cette approximation est que la molécule n'est pas rigide, et ses vibrations sont complètement harmoniques. En conséquence, en réalité, nous avons besoin de l'approximation d'un rotateur non rigide et d'un oscillateur anharmonique. Et le mot clé ici est «anharmonique», c'est-à-dire "Pas harmonique."Parlons des molécules les plus simples: diatomiques. Il en existe de nombreux exemples: HCl, HBr, HI, CO, O 2 , N 2 , etc. etc.
Elles se distinguent de toutes les molécules par le fait qu'elles n'ont qu'une seule vibration: l'extension / compression de la distance interatomique.Et c'est la distance entre les atomes que l'on peut mesurer en diffraction d'électrons gazeux (dans une variante de la température moyenne,r g ) et en spectroscopie rotationnelle (moyenne sur, disons, l'état vibratoire du sol, c'est-à-direr 0 )
Et maintenant, la principale question se pose de l'univers de la vie et en général:quel sera r g etr 0 dans l'approximation d'un oscillateur harmonique, et comment cela se corrèle-t-il avec la distance d'équilibrer e ?
Pour une réponse, il faut regarder la surface d'énergie potentielle d'une molécule diatomique:
- , : . , .
- , , 2 :
- ( r→0 ), - , « » ,
- ( r→+∞ )
En conséquence, si la molécule raccourcit sa longueur de liaison par rapport à la position d'équilibre, elle bute contre le mur, et si elle augmente, elle tombe sur un canapé moelleux. Et la molécule n'est pas un imbécile, elle reposera plus sur le canapé que sur le mur. Par conséquent, la vibration moyenne sur la distance sera supérieure à l'équilibre (r e < r 0 , r g ), et cela se remarque: ces déplacements sont de l'ordre de 0,01 Å, ce qui est supérieur aux erreurs de mesure.
Par conséquent, même si nous voulons calculer quelque chose de plus comme une expérience, en restant dans le cadre du modèle de molécule standard (RR-HO @ BO), nous n'obtiendrons rien de nouveau, par conséquent, la géométrie même à l'équilibre participera à la comparaison.Conclusion d'erreur # 2
 Illustration de l'article DOI: 10.1002 / anie.201611308 .Et la conclusion est terriblement simple et se compose de 2 parties.
Illustration de l'article DOI: 10.1002 / anie.201611308 .Et la conclusion est terriblement simple et se compose de 2 parties.- Avec une comparaison correcte, toutes les valeurs doivent avoir la même signification.
- Si les valeurs sont différentes, cela ne doit pas être oublié.
Exemples d'erreurs dans des articles scientifiques
"Oeuvres hindoues"
En fait, le principal endroit où vous pouvez trouver ce sont les magazines de bas niveau. Ils contiennent rarement des articles avec des résultats sympas, ils ont donc été choisis par les «principaux chercheurs» des pays du deuxième monde et plus (les pays BRICS et leurs adeptes moins réussis). Par magazines de «bas niveau», on entend ici non pas ceux qui publient des articles tels que «The Rooter: algorithme pour l'unification typique des points d'accès et de la redondance», mais plutôt des publications scientifiques respectées. Dans mon domaine scientifique, les «demi-lavages» les plus connus sont:(il y en a d'autres). Comme vous pouvez le voir, selon des signes officiels, dans la science russe, ils sont considérés comme des publications très décentes. Mais, il y a un tel afflux de contenu r ... de qualité douteuse que beaucoup fuit encore.À titre d'illustration, j'ai pris le dernier numéro du Journal of Molecular Structure et parcouru la table des matières, et le tour est joué:S. Sathiya, M. Senthilkumar, C. Ramachandra Raja, Croissance cristalline, analyse de surface Hirshfeld, étude DFT et études NLO de troisième ordre de la thiourée 4 diméthyl aminobenzaldéhyde // J. Mol. Struct., V. 1180 (2019), PP. 81-88.
https://doi.org/10.1016/j.molstruc.2018.11.067
La structure générale d'un tel travail est très sans prétention.- Une substance est «bouillie» (mais le plus souvent achetée bêtement sur Sigma ). Dans ce travail, la substance est encore cuite.
- (), . — , ( , ) , . , - Gaussian, « ». … 1 2 , .. - .
- , /Vis, .
- Dans les visualiseurs moléculaires standard, tels que GaussView (grogné), de belles images sont dessinées, mais toujours de mauvaise qualité.
- Aucune conclusion de fond n'est tirée: «nous avons beaucoup expérimenté, beaucoup compté, apporté des tableaux et des images ⇒ nous sommes grands, donne-nous des bonbons. »
Mais de mes favoris: articleM. Govindarajan, M. Karabacak, FT-IR, FT-Raman et investigation spectrale UV; analyse d'estimation de fréquence calculée et calculs de structure électronique sur 1-nitronaphtalène // Spectrochimica Acta Partie A: Spectroscopie moléculaire et biomoléculaire, V. 85 (2012), PP. 251-260,
https://doi.org/10.1016/j.saa.2011.10.002.
Dans ce document, le spectre en phase solide a été stupidement enregistré, puis interprété sur la base de calculs médiocres dans le modèle HO en phase gazeuse. Mais l'astuce est qu'ils ne pouvaient même pas faire de calculs normaux,
ce qu'ils ont poliment laissé entendre dans le commentaire de l'article .
Cependant, le terme "œuvres hindoues" (comme le terme "
code hindou ") fait référence loin aux seules œuvres provenant du sous-continent mystérieux correspondant.
Si vous allez sur le merveilleux site de la
Cyberleninka , alors
vous regardez les Abysses, et les Abysses vous regardent, vous pouvez découvrir beaucoup de choses intéressantes. En recherchant «chimie quantique» (avec / sans la condition supplémentaire «rsa»), il a été possible de trouver de nombreuses choses utiles. Puisqu'une grande partie des travaux «chimie quantique» était consacrée à l'étude des chevaux sphériques dans le vide (c'est-à-dire des calculs sans référence à la réalité), ils n'étaient pas liés à ce texte. Mais parmi eux, ces trois œuvres ont été perdues:
J'ai été particulièrement satisfait de ce dernier, car «l'évaluation de l'adéquation» était une comparaison inadéquate des structures dans différentes phases (gaz vs cristal) et avec des significations différentes (
r e contre
r a l p h a ) - c'est vraiment le comble de l'adéquation.
Cela arrive-t-il dans les bons magazines?
Oui, il y a des «erreurs».
N'ayez pas peur, tout s'est bien terminé!Heureusement, dans l'histoire à laquelle nous allons maintenant nous tourner, une attitude négligente envers les valeurs expérimentales / théoriques n'a pas entraîné de conséquences graves et terribles. Et l'œuvre, malgré la véritable omission, ne perd pas son sang-froid et sa signification.
De plus, au final, tout s'est terminé en super général: la publication d'un article génial où la justice a triomphé.
Nous parlons d'une des molécules organiques avec une liaison C - C extrêmement longue: le
1,1'-bisdiadamantane :

Pourquoi cette molécule est coolSi nous regardons la chimie scolaire ou
les manuels
universitaires en chimie, sur
Wiki (ou
même simplement sur Google ), nous découvrirons que la longueur standard d'une seule liaison C-C est de 1,54 Å.
Ainsi, la longueur expérimentale de la simple liaison centrale dans le 1,1'-bisdiadamantane
re=1,630 pm0,005 Å, presque 0,08 Å de plus (c'est du dofiga)!
Cette extension d'une seule liaison C - C se produit parce que les deux pièces semblables à des diamants que cette liaison détient sont très grandes, elles se repoussent donc. Mais, comme nous nous en souvenons de l'exemple du Ferrocène, nous avons également une attraction due aux forces de dispersion (Londres). Et en raison de la taille maladive de ces moitiés de la molécule, il existe une grande attraction de dispersion entre les pièces. Il ne permet pas à cette liaison centrale de se rompre, de sorte que cette molécule peut (relativement facilement) être vaporisée sans la rompre en cours de route.
La connexion unaire centrale C - C est très longue, et il était donc très intéressant de comparer la façon dont les méthodes théoriques reproduisent la réalité. Et la réalité est dans les premiers articles, les voici:
Putain de merde!Le premier article est dans Nature, le magazine le plus cool, et le second dans JACS, le magazine purement chimique le plus respecté !!! Peu de gens en rêvaient, mais cette collaboration scientifique germano-ukrainienne est très cool.
n'était représentée que par des données cristallographiques. En conséquence, en comparant des choses comparables difficiles entre elles, sans apporter de corrections
appropriées à la différence de paramètres, ils ont finalement conclu que la longueur de la liaison centrale C-C dans le gaz est de 1,655 Å, dépassée de 0,02 Å. Et c'est bien plus que l'erreur expérimentale.
Heureusement, à la fin, ils ont collaboré avec des spécialistes sur ces questions
et ont finalement reçu la bonne réponse (un
court résumé populaire de ce travail peut également être trouvé sur N + 1 ).
Avez-vous besoin d'une comparaison?

Après tout ce que j'ai écrit sur l'exactitude des comparaisons, une question raisonnable peut se poser: faut-il alors comparer entre eux les résultats des calculs et les résultats des expériences?
C'est nécessaire! Tout comme vous en avez besoin!Il y a une déclaration célèbre (dont je n'ai pas pu trouver un auteur fiable):
Personne ne croit aux calculs théoriques, sauf celui qui les a faits.
Tout le monde croit aux résultats expérimentaux, sauf celui qui les a obtenus.
Traduit en russe, cela ressemble à ceci: personne ne croit aux calculs théoriques, sauf celui qui les a faits, mais tout le monde croit aux données expérimentales, sauf celui qui les a reçues.
Mais en science, il faut que tout le monde croie (enfin, ou la majorité), et l'expérience est la seule mesure, car elle relie ce que nous avons calculé à la réalité.
Il y a un remarquable essai (en accès libre) dans la deuxième revue chimique la plus importante sur le sujet de ce qu'un chercheur ou une personne moyenne lisant des articles scientifiques et / ou des nouvelles devrait faire:
Mata R., Suhm M. // Angew. Chem. Int. Ed., 56 (2017), DOI: 10.1002 / anie.201611308(au fait, j'y ai déjà donné un lien, car une photo, par Ricardo Mata, est de cet article).
Les conclusions de cet essai fournissent des recommandations aux théoriciens et expérimentateurs de la simulation. Je vais les donner ici (en traduction et une petite révision) comme un dernier mot à ce post.
- Le théoricien doit:
- donner non seulement des méthodes et des tentatives réussies, mais aussi décrire les échecs des méthodes (surtout si ces méthodes sont populaires),
- décrivez bien et entièrement votre méthodologie,
- où il est possible de donner des estimations (ou une description) des erreurs et d'importantes approximations et simplifications acceptées.
- Les expérimentateurs, à leur tour, doivent:
- pour pousser les théoriciens à leurs données expérimentales, qu'ils pourraient utiliser comme références (normes),
- montrer à la communauté scientifique des données expérimentales incompréhensibles, où la théorie (ou des expériences supplémentaires) aideraient à l'explication,
- parler en
territoire étranger de conférences théoriques avec leurs données,
histoire personnelleSoit dit en passant, je viens de rencontrer le deuxième auteur de cet article, qui est un expérimentateur, lors d'un colloque théorique.
- retirer de vos données expérimentales des éléments aussi accessibles que possible pour la comparaison avec la théorie,
Toutes les comparaisons les meilleures et correctes! Et souvenez-vous: seul celui qui ne fait rien ne se trompe pas.
PS
En guise de postface, je voudrais donner une petite liste de bases de données où vous pouvez rechercher diverses données expérimentales pour les molécules.
Banques de données structurelles
Boîtes cristallographiques
La façon la plus simple de déterminer la structure d'une molécule dans un cristal est L'ACP est une routine. Par conséquent, si vous ne savez pas à quoi ressemble la molécule, allez aux banques de données cristallographiques (des endroits où presque toutes les structures de substances qui ont été entassées dans un goniomètre et illuminées par un faisceau de particules à ondes courtes sont collectées). Puisqu'il y a beaucoup de telles banques, je ne donnerai que les plus célèbres (une liste plus complète peut être trouvée sur le
Wiki ).
- Base de données sur la structure des cristaux inorganiques ( ICSD ). Il ne s'agit pas uniquement de molécules, vous y trouverez principalement des structures de différents sels, métaux, céramiques, etc. Cette base est prise en charge par l'Université de technologie de Karlsruhev, donc son accès est payant et pas bon marché. Mais si ça, son site .
- Cambridge Structural Database ( CSD ). Peut-être la plus grande base cristallographique du monde. Près d'un million de structures! Ce sont principalement des cristaux moléculaires organiques et organométalliques. Et cette base de données est gratuite! Pour ajouter. des frais, bien sûr, vous pouvez obtenir une option intéressante, comme une recherche sur la structure dessinée de la molécule, mais cela est déjà excédentaire. Le site .
- Base de données ouverte de cristallographie ( COD ). Eh bien, aussi une sorte de base de données. Ce qui est là, je ne sais pas vraiment, mais au moins ça existe. Le site .
- Eh bien, pour ceux qui s'intéressent à la biologie, il existe une banque de données sur les protéines ( PDB ). Une base de données ouverte où vous pouvez télécharger les structures de protéines énormes et effrayantes (mais en cristaux). Le site .
La structure des molécules dans un gaz
Ici, les choses sont un peu pires, car les expériences pour étudier la structure des molécules libres sont beaucoup plus compliquées, tant pour la réalisation (au moins un vide poussé est nécessaire) que pour l'interprétation.
Par conséquent, il y a beaucoup moins de bases de données.
- La plus grande base de données pour les molécules libres est la DOCumentation MOlecular GAsphase (ou MOGADOC ). Il est basé à l'Université d'Ulm et représente un investissement très coûteux. Mais, si quoi que ce soit, le site est ici .
- Si vous voulez connaître les structures d'équilibre expérimentales à 100% des molécules, il s'agit de la base de données NIST Computational Chemistry Comparison and Benchmark DataBase ( CCCBDB ). Presque tous purement expérimentaux r e -les structures peuvent être trouvées là-bas, mais il y a suffisamment d'autres nishtyaks là-bas. Le site .
- Le dernier bastion est le projet sous - développé de MolWiki . Mais il n'y a pas beaucoup de structures qui s'y trouvent, seulement celles qui ont été reçues à l'Université de Bielefeld, à l'Université d'État de Moscou et à l'ISTU ces dernières années (et même pas toutes). Le site .
Où trouver les caractéristiques spectrales des molécules?
 Tiré de xkcd.com
Tiré de xkcd.comIl y a beaucoup plus de bases de données ici, car supprimer le spectre sans interprétation est une tâche beaucoup plus simple que d'obtenir une structure (pas besoin de construire des modèles et de prouver qu'ils sont corrects). De plus, les spectres ont une grande valeur appliquée: ils peuvent être utilisés pour déterminer la composition des échantillons, qu'il s'agisse d'eau d'une rivière voisine ou d'un signal provenant d'un nuage moléculaire (ou même d'une atmosphère d'exoplanète, voir l'illustration ci-dessus).
Oui, d'ailleurs, tous les liens de cette section seront vers des bases de données gratuites.
- NIST Chemistry WebBook . Peut-être le meilleur livre du monde. Dans cette base de données, vous pouvez trouver IR, UV / Vis, spectres de masse pour un tas de molécules. Et aussi, des paramètres thermochimiques et même parfois cinétiques! En général, si un chimiste ne connaît pas l'existence de ce site, il n'est pas chimiste. Le site .
- Base de données d'absorption moléculaire de transmission à haute résolution ( HITRAN ). Le projet du Harvard-Smithsonian Center for Astrophysics, dont il est évident que les spectres qui y sont montrés sont utilisés pour identifier des molécules dans des espaces interstellaires et circumstellaires (par exemple, comme celui-ci ). Le site .
- Semblable au précédent, le projet The Cologne Database for Molecular Spectroscopy, de la partie astrophysique de l'Université de Cologne. Le site .
- Eh bien, comme dernier exemple, je vais donner le projet ExoMol . Ce ne sont pas des spectres expérimentaux entièrement purs, mais c'est un excellent exemple de l'interaction de la théorie et de l'expérience: sur la base de données expérimentales de haute précision et de calculs de très haut niveau, les spectres de molécules simples sont prédits dans des conditions différentes (y compris extrêmes). L'accent principal est mis ici sur les biomarqueurs, donc lorsque les astronomes voient le spectre des exoplanètes, ils peuvent facilement identifier les molécules que nous connaissons déjà en eux. Le site .
PPS
S'il y a des erreurs / quelque chose reste incompréhensible, écrivez dans les commentaires - je vais corriger / essayer de mieux expliquer.