Il n'y a pas si longtemps, j'ai lu le roman de science-fiction
«Le problème des trois corps» de Liu Qixin . Certains extraterrestres y avaient un problème - ils ne savaient pas comment, avec une précision suffisante pour eux, calculer la trajectoire de leur planète d'origine. Contrairement à nous, ils vivaient dans un système à trois étoiles, et le «temps» sur la planète dépendait fortement de leur emplacement relatif - de la chaleur incinérante au froid glacial. Et j'ai décidé de vérifier si nous pouvons résoudre de tels problèmes.
Physique du phénomène
Pour comprendre le problème, il est nécessaire de traiter la physique du phénomène. Dans le cadre de la théorie classique, la force d'attraction de deux corps est déterminée par
la loi
de Newton :
v e c F ( v e c r 1 , v e c r 2 ) = - G m 1 m 2 f r a c v e c r 1 - v e c r 2 | v e c r 1 - v e c r 2 | 3 ,
où
vecr1, vecr2 - la position des corps dans l'espace,
m1,m2 - masses de corps,
G - constante gravitationnelle.
Dans le système de
N Les corps sur chacun d'eux seront affectés par la force d'attraction du reste, qui est exprimée par l'équation:
vecFn=−G sumk neqnmnmk frac vecrn− vecrk| vecrn− vecrk|3.
En utilisant
la deuxième loi de Newton, nous écrivons l'accélération pour chaque particule:
vecan= vecFn/mn=−G sumk neqnmk frac vecrn− vecrk| vecrn− vecrk|3.
Rappelant que l'accélération est la dérivée seconde du temps de la coordonnée, nous obtenons une équation différentielle partielle de second ordre, qui doit être résolue pour obtenir la trajectoire de chaque corps:
frac partial2 vecrn partialt2=fn=−G sumk neqnmk frac vecrn− vecrk| vecrn− vecrk|3.
Il est important de noter ici que la complexité du calcul d'une fonction
fn est égal à
O(N2) et augmente de manière significative avec l'augmentation du nombre de corps en interaction.
Math
La première et la plus simple méthode pour résoudre des équations différentielles est
la méthode d'Euler , qui est conçue pour résoudre des équations de la forme:
fracdydx=f(x,y).
Lors de la transition vers la région discrète, nous obtenons:
yi=yi−1+hf(xi−1,yi−1), quadi=1,2,3, dots,m,
où
h Est l'étape d'intégration, et
m - le nombre d'étapes d'intégration. Ainsi, si nous devons calculer la position des corps à la fois
T alors on devrait faire
m=T/h étapes d'intégration. Ici, le premier problème est visible - si
T grand, alors nous devons prendre un grand nombre d'étapes d'intégration.
Pour appliquer la méthode d'Euler à notre problème, elle doit être réduite à un système de premier ordre. Pour ce faire, nous introduisons une variable supplémentaire - la vitesse des particules:
frac partial vecvn partialt=fn, frac partial vecrn partialt= vecvn.
Le deuxième problème dans la résolution de systèmes d'équations différentielles est la précision de la solution et son contrôle. La précision peut être améliorée de deux manières: en diminuant l'étape d'intégration et en choisissant une méthode avec un ordre de précision plus élevé. Les deux méthodes conduisent à une complexité de calcul accrue, mais de différentes manières. Par exemple, vous pouvez utiliser la
méthode Runge-Kutta classique du
quatrième ordre , elle nécessite quatre calculs de fonction
fn à chaque étape, mais a un ordre de précision
O(h4) (à titre de comparaison, la méthode Euler a un ordre de précision
O(h) et nécessite un calcul
fn ) La précision de la solution peut également être contrôlée de plusieurs manières: comparer avec la solution analytique, résoudre en utilisant différentes méthodes ou avec différentes étapes et comparer les résultats, contrôler les paramètres tiers et les contraintes auxquelles la solution doit se conformer.
De plus, chacune de ces méthodes a ses inconvénients. Les décisions analytiques peuvent être absentes ou, même dans la plupart des cas, complètement absentes. Par exemple, pour notre tâche
N Cette solution analytique est
N=2 , mais même cela suffit pour tester la précision des méthodes. La résolution d'un problème par deux méthodes ou avec des étapes différentes augmente le temps de calcul, mais cette approche peut être appliquée à presque toutes les tâches. Toutes les tâches n'ont pas de limites, mais pour la nôtre, elles les ont: à chaque étape de l'intégration, nous pouvons contrôler la mise en œuvre
des lois de conservation . Cette approche augmente également la complexité du calcul, mais il y a beaucoup de choix; le calcul de la somme des impulsions ou des impulsions angulaires de toutes les particules a une complexité de l'ordre
O(N) , tout en calculant l'énergie totale d'un système a une complexité d'ordre
O(N2)Remarque sur le calcul de l'énergie totaleDans notre cas, l'énergie totale du système se compose de deux parties - l'énergie cinétique et l'énergie potentielle. L'énergie cinétique consiste en la somme des énergies cinétiques de tous les corps. Pour calculer l'énergie potentielle, nous devons ajouter les énergies potentielles de chaque particule dans le champ gravitationnel des particules restantes, nous devons donc ajouter
O(N2) termes. La difficulté est que tous les termes sont d'un ordre très différent, et même avec des calculs en double précision, il n'est pas possible de calculer cette valeur avec une précision suffisante pour la comparaison à différentes étapes. Pour surmonter ce problème, il a fallu appliquer la sommation selon
l'algorithme de Kahan .
Fig 1. Un exemple de trajectoire elliptique.
Prenons le cas simple d'un satellite se déplaçant sur une orbite elliptique autour de la Terre. À mesure que le satellite s'approche de la Terre, sa vitesse augmente et, en s'éloignant de la Terre, il diminue, en conséquence, la possibilité se présente de diminuer le pas d'intégration dans le temps dans la partie verte de l'orbite et de l'augmenter en rouge et bleu sans changer la précision de la solution. Essayons de comparer plus en détail.
Tableau 1. Méthodes étudiées pour résoudre les équations différentiellesPour sélectionner la meilleure méthode pour notre tâche, nous comparerons plusieurs méthodes connues. Pour ce faire, nous simulons la collision de deux systèmes de corps
N=512 et mesurer la variation relative de l'
énergie totale , de la
quantité de mouvement et de son
moment à la fin de la simulation (temps de simulation maximum
Tmax=2,5 ) Dans ce cas, nous varierons l'étape et les paramètres des méthodes d'intégration et mesurerons le nombre d'appels de fonction
fn , respectivement, les méthodes qui avec un plus petit nombre d'appels conduiront à moins de pertes, nous considérerons plus acceptables.

| 
|
| a) | b) |
| Figure 2. Variation relative de l'énergie a), momentum b), à la fin de la simulation du système N=512 corps par diverses méthodes en fonction du nombre de calculs de fonction fn double précision |
À partir des graphiques de la figure 2, on peut voir que le meilleur rapport de la quantité de calcul de la fonction
fn et le changement relatif de l'énergie du système des corps dans les méthodes d'Adams et Dorman-Prince du cinquième ordre. On voit également que pour toutes les méthodes avec un nombre croissant de calculs
fn la variation relative de la dynamique du système augmente. Pour un changement relatif d'énergie, cela est également perceptible, mais seulement pour quelques méthodes qui pourraient atteindre le seuil
dE/E0<10−12 . Cet effet ne peut plus être attribué à l'erreur des méthodes, mais à l'erreur des calculs, et une nouvelle augmentation de la précision n'est possible qu'avec une augmentation de la précision des calculs à virgule flottante.

| 
|
| a) | b) |
| Fig. 3. Variation relative de l'énergie a), momentum b), à la fin de la simulation du système N=512 corps par diverses méthodes en fonction du nombre de calculs de fonction fn avec une quadruple précision (__float128) |
Les figures 3a et 3b montrent que l'utilisation de calculs avec une précision quadruple peut réduire les pertes d'énergie relatives jusqu'à

, mais vous devez comprendre que le temps de calcul est augmenté de deux ordres de grandeur par rapport à la double précision. Comme dans le cas des calculs en double précision, le meilleur rapport de précision et le nombre de calculs de fonction
fn posséder des méthodes du cinquième ordre Adams et Dorman-Prince.
Les
méthodes Dorman-Prince et Werner appartiennent à la classe des
méthodes imbriquées et peuvent calculer simultanément deux solutions avec un ordre de précision élevé et faible (pour la méthode Dorman-Prince, ordres 5 et 4, et pour la méthode Werner, ordres 6 et 5). Si ces deux solutions sont très différentes, nous pouvons diviser l'étape d'intégration actuelle en plus petites. Cela nous permet de changer dynamiquement l'étape d'intégration et de la réduire uniquement dans les domaines où cela est nécessaire.
Comparons plus en détail les méthodes Dorman-Prince, Werner et Adams du cinquième ordre sur un intervalle de simulation plus long de notre système (
Tmax=300 )

|
a) Le changement relatif d'énergie, de quantité de mouvement et son moment dans le processus de modélisation
|

|
b) Nombre de calculs de fonction fn sur l'intervalle de simulation DeltaT=0,3
|
Fig. 4. Dépendances de la variation relative des paramètres physiques du système et du nombre de calculs de fonction fn modélisation ponctuelle avec la méthode à pas variable de Dorman-Prince h=10−5 points10−9
|

|
a) Le changement relatif d'énergie, de quantité de mouvement et son moment dans le processus de modélisation
|

|
b) Nombre de calculs de fonction fn sur l'intervalle de simulation DeltaT=0,3
|
Fig. 5. Dépendances de la variation relative des paramètres physiques du système et du nombre de calculs de fonction fn par rapport à la modélisation temporelle par la méthode de Werner avec un pas variable h=10−5 points10−9
|

|
Fig. 6. Changement relatif d'énergie, d'élan et de son moment dans le processus de modélisation par la méthode d'Adams-Bashfort du cinquième ordre avec une étape h=10−6
|

|
Fig 7. Dépendances du nombre de calculs de fonction fn pour les méthodes Adams, Dorman-Prince et Werner du cinquième ordre à partir du temps de simulation
|
On peut voir qu'au stade initial de l'évolution de notre système (
T<25 ), les trois méthodes présentent des caractéristiques similaires, mais à des stades ultérieurs, certains événements se produisent dans le système, à la suite desquels les erreurs dans les principaux paramètres du système (énergie totale, quantité de mouvement et sa quantité de mouvement) augmentent fortement. Mais les méthodes Dorman-Prince et Werner font face à ces changements beaucoup mieux en raison de la capacité à réduire l'étape d'intégration dans les sections «complexes», ce qui augmente le nombre de calculs de fonctions
fn comme le montrent les figures 4b et 5b, mais le nombre total de calculs
fn les méthodes imbriquées sont plus petites que la méthode Adams-Bashfort, comme le montre la figure 7.
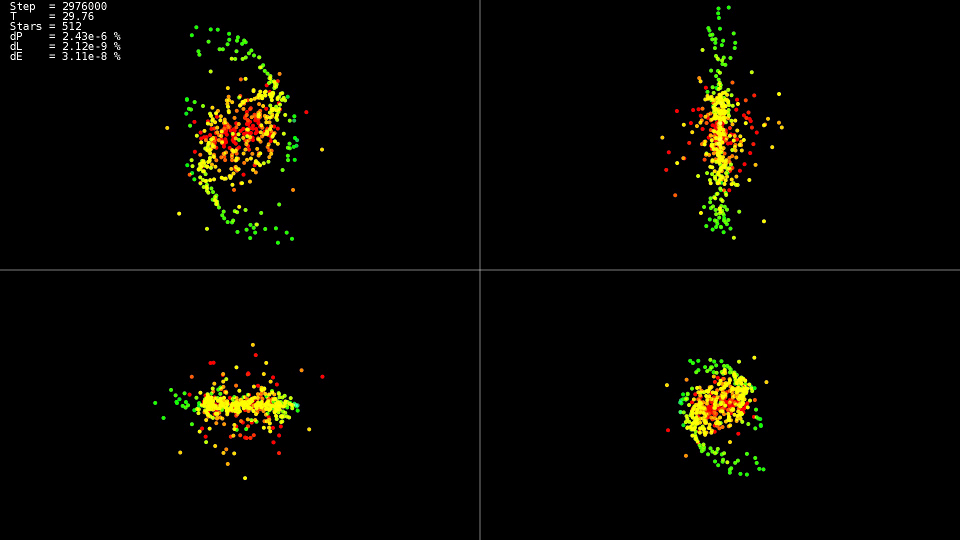
Je me demande ce qui est arrivé au système à ces moments
|
Vidéo 1. Modélisation d'un système de 512 corps. Méthode Dorman-Prince. Étape dynamique h=10−5 points10−9
|
La vidéo montre que jusqu'au point dans le temps
T=25 le mouvement est relativement calme, et après une collision des centres des «galaxies» se produit, ce qui entraîne un changement brusque des trajectoires et une forte augmentation des vitesses de certaines particules. De plus, pour maintenir la précision de la solution, il est nécessaire de réduire l'étape d'intégration. Les méthodes imbriquées peuvent le faire automatiquement; les graphiques montrent que dans certaines parties de l'évolution du système, l'étape d'intégration a été réduite de près de deux ordres de grandeur avec

avant
h=10−7 . En utilisant la méthode Adams et une telle étape sur tout l'intervalle de l'évolution du système, nous n'aurions pas attendu de solution.
Résumé
Pour la solution, il est préférable d'utiliser des méthodes imbriquées qui vous permettent de contrôler dynamiquement l'étape d'intégration et de la réduire uniquement sur des sections "complexes" de la trajectoire.
Ne poursuivez pas les méthodes les plus élevées. Même lorsqu'ils utilisent le type de données «double», ils n'atteignent pas leurs capacités potentielles, l'utilisation des types de données avec une plus grande précision augmente considérablement le temps nécessaire pour résoudre le problème.
Implémentation CPU
Maintenant que le choix d'une méthode de résolution d'équations est défini, essayons de comprendre le calcul de la force d'interaction pour chaque particule. Nous obtenons un double cycle pour toutes les particules:
Code de mise en œuvre «simple»for(size_t body1 = 0; body1 < count; ++body1) { const nbvertex_t v1(rx[ body1 ], ry[ body1 ], rz[ body1 ]); nbvertex_t total_force; for(size_t body2 = 0; body2 != count; ++body2) { if(body1 == body2) { continue; } const nbvertex_t v2(rx[ body2 ], ry[ body2 ], rz[ body2 ]); const nbvertex_t force(m_data->force(v1, v2, mass[body1], mass[body2])); total_force += force; } frx[body1] = vx[body1]; fry[body1] = vy[body1]; frz[body1] = vz[body1]; fvx[body1] = total_force.x / mass[body1]; fvy[body1] = total_force.y / mass[body1]; fvz[body1] = total_force.z / mass[body1]; }
Les forces gravitationnelles pour chaque corps sont calculées indépendamment, et pour utiliser tous les cœurs de processeur, il suffit d'écrire la directive OpenMP avant le premier cycle:
Un morceau de code de la mise en œuvre de «openmp» #pragma omp parallel for for(size_t body1 = 0; body1 < count; ++body1)
Parce que chaque corps interagit avec chacun, puis pour réduire le nombre d'interactions du processeur avec la RAM et améliorer l'utilisation du cache, nous avons la possibilité de charger une partie des données dans le cache et de l'utiliser à plusieurs reprises:
Code d'implémentation 'openmp + block' #pragma omp parallel for for(size_t n1 = 0; n1 < count; n1 += BLOCK_SIZE) { nbcoord_t x1[BLOCK_SIZE]; nbcoord_t y1[BLOCK_SIZE]; nbcoord_t z1[BLOCK_SIZE]; nbcoord_t m1[BLOCK_SIZE]; nbvertex_t total_force[BLOCK_SIZE]; for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; x1[b1] = rx[local_n1]; y1[b1] = ry[local_n1]; z1[b1] = rz[local_n1]; m1[b1] = mass[local_n1]; } for(size_t n2 = 0; n2 < count; n2 += BLOCK_SIZE) { nbcoord_t x2[BLOCK_SIZE]; nbcoord_t y2[BLOCK_SIZE]; nbcoord_t z2[BLOCK_SIZE]; nbcoord_t m2[BLOCK_SIZE]; for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { size_t local_n2 = b2 + n2; x2[b2] = rx[local_n2]; y2[b2] = ry[local_n2]; z2[b2] = rz[local_n2]; m2[b2] = mass[n2 + b2]; } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { const nbvertex_t v1(x1[ b1 ], y1[ b1 ], z1[ b1 ]); for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { const nbvertex_t v2(x2[ b2 ], y2[ b2 ], z2[ b2 ]); const nbvertex_t force(m_data->force(v1, v2, m1[b1], m2[b2])); total_force[b1] += force; } } } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; frx[local_n1] = vx[local_n1]; fry[local_n1] = vy[local_n1]; frz[local_n1] = vz[local_n1]; fvx[local_n1] = total_force[b1].x / m1[b1]; fvy[local_n1] = total_force[b1].y / m1[b1]; fvz[local_n1] = total_force[b1].z / m1[b1]; } }
Une optimisation supplémentaire consiste à supprimer le contenu de la fonction de calcul de la force dans le cycle principal et d'éliminer la division et la multiplication par la masse corporelle m1 [b1]. Outre le fait que nous avons un peu réduit les calculs, le compilateur pourra appliquer des instructions vectorielles des processeurs SSE et AVX sur un cycle aussi étendu.
Code d'implémentation 'openmp + bloc + optimisation' #pragma omp parallel for for(size_t n1 = 0; n1 < count; n1 += BLOCK_SIZE) { nbcoord_t x1[BLOCK_SIZE]; nbcoord_t y1[BLOCK_SIZE]; nbcoord_t z1[BLOCK_SIZE]; nbcoord_t total_force_x[BLOCK_SIZE]; nbcoord_t total_force_y[BLOCK_SIZE]; nbcoord_t total_force_z[BLOCK_SIZE]; for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; x1[b1] = rx[local_n1]; y1[b1] = ry[local_n1]; z1[b1] = rz[local_n1]; total_force_x[b1] = 0; total_force_y[b1] = 0; total_force_z[b1] = 0; } for(size_t n2 = 0; n2 < count; n2 += BLOCK_SIZE) { nbcoord_t x2[BLOCK_SIZE]; nbcoord_t y2[BLOCK_SIZE]; nbcoord_t z2[BLOCK_SIZE]; nbcoord_t m2[BLOCK_SIZE]; for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { size_t local_n2 = b2 + n2; x2[b2] = rx[local_n2]; y2[b2] = ry[local_n2]; z2[b2] = rz[local_n2]; m2[b2] = mass[n2 + b2]; } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { nbcoord_t dx = x1[b1] - x2[b2]; nbcoord_t dy = y1[b1] - y2[b2]; nbcoord_t dz = z1[b1] - z2[b2]; nbcoord_t r2(dx * dx + dy * dy + dz * dz); if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = (m2[b2]) / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; total_force_x[b1] -= dx; total_force_y[b1] -= dy; total_force_z[b1] -= dz; } } } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; frx[local_n1] = vx[local_n1]; fry[local_n1] = vy[local_n1]; frz[local_n1] = vz[local_n1]; fvx[local_n1] = total_force_x[b1]; fvy[local_n1] = total_force_y[b1]; fvz[local_n1] = total_force_z[b1]; } }
Tableau 2. Dépendance du temps de calcul (en secondes) d'une fonction fn sur le nombre de corps en interaction N pour diverses implémentations CPU| N | 2048 | 4096 | 8192 | 16384 | 32768 |
|---|
| simple | 0,0425 | 0.1651 | 0,6594 | 2,65 | 10,52 |
| openmp | 0,0078 | 0,0260 | 0,1079 | 0,417 | 1,655 |
| openmp + bloc + optimisation | 0,0037 | 0,0128 | 0,0495 | 0,194 | 0,774 |
Paramètres système:- système: Debian 9, Intel Core i7-5820K (6 cœurs)
- compilateur: gcc 6.3.0
On voit clairement que la version avec prise en charge d'OpenMP est accélérée six fois, exactement en termes de nombre de cœurs, et la version optimisée est plus rapide d'un peu plus de deux fois. Ainsi, avec l'optimisation, vous ne devez pas vous fier uniquement à la concurrence. Fait intéressant, dans les calculs à flux unique, le processeur fonctionnait à une fréquence de 3,6 GHz, dans la version parallèle (openmp), il a baissé la fréquence à 3,4 GHz, et dans le parallèle et optimisé (openmp + bloc + optimisation), il est tombé à 3,3 GHz, mais cela cela ne l'a pas empêchée de travailler 13,6 fois plus vite. On voit également qu'une augmentation du temps de calcul avec une augmentation de la taille du problème est quadratique, et une augmentation supplémentaire
N rend la tâche insoluble dans un délai raisonnable.
Implémentation GPU
Mais il y a un désir de faire des calculs encore plus rapidement. Il existe plusieurs directions disponibles pour l'accélération: calcul GPU, approximation des fonctions
fn . Tout d'abord, pour l'informatique GPU, j'ai choisi la technologie OpenCL. Pour un travail plus pratique, la bibliothèque
CLHPP a été utilisée. Le principal avantage d'OpenCL est que le code peut être exécuté sur le processeur et sur le GPU, ce qui simplifie l'écriture et le débogage, ainsi que l'extension de la liste du matériel à exécuter. L'outil
Oclgrind aide au débogage qui, lors de l'exécution, affiche des appels d'API OpenCL incorrects et des problèmes d'accès à la mémoire.
Opencl
Pour commencer avec OpenCL, vous devez obtenir une liste des plates-formes disponibles. Les plates-formes les plus courantes sont AMD, Intel et NVidia.
Code std::vector<cl::Platform> platforms; cl::Platform::get(&platforms);
Ensuite, après avoir choisi une plate-forme, vous devez sélectionner le périphérique informatique que cette plate-forme représente:
Code const cl::Platform& platform(platforms[platform_n]); std::vector<cl::Device> all_devices; platform.getDevices(CL_DEVICE_TYPE_ALL, &all_devices);
Et à la fin de la phase préparatoire, il est nécessaire de créer un contexte et des files d'attente dans lesquels la mémoire sera allouée et les calculs seront effectués. Par exemple, un contexte qui combine tous les appareils informatiques d'une plate-forme sélectionnée est créé comme suit:
Code de création de contexte et de file d'attente cl::Context context(all_devices); std::vector<cl::CommandQueue> queues; for(cl::Device device: all_devices) queues.push_back(cl::CommandQueue(context, device));
Pour télécharger le code source sur un appareil informatique, il doit être compilé, la classe cl :: Program est destinée à cela.
Code de compilation du noyau std::vector< std::string > source_data; cl::Program::Sources sources; for(int i = 0; i != files.size(); ++i) { source_data.push_back(load_program(files[i]));
Pour décrire les paramètres d'une fonction (noyau) exécutée sur un appareil informatique, il existe un modèle cl: make_kernel.
Exemple de noyau de calcul de la force d'interaction typedef cl::make_kernel< cl_int, cl_int,
De plus, tout est simple: on déclare une variable avec le type du noyau, on y passe le programme compilé et le nom du noyau de calcul, on peut démarrer le noyau presque comme une fonction normale.
Code de lancement du noyau ComputeBlock fcompute(prog, "ComputeBlockLocal"); cl::NDRange global_range(device_data_size); cl::NDRange local_range(block_size); cl::EnqueueArgs eargs(ctx.m_queue, global_range, local_range); fcompute(eargs, ... );
Le noyau de calcul pour OpenCL lui-même est très similaire à l'option 'openmp + block + optimisation' pour le CPU, seulement contrairement à la version du CPU, le premier cycle est contrôlé à l'aide d'OpenCL (la plage de cycle est déterminée par la variable global_range du code de démarrage du noyau, et le numéro d'itération actuel est disponible à partir du noyau en utilisant la fonction get_global_id (0)). Tout d'abord, une partie des données du corps est chargée à partir de la mémoire locale, puis traitée. La mémoire locale étant commune à tous les threads du groupe, le téléchargement a lieu une fois pour le groupe et est traité par chaque thread du groupe et, depuis la mémoire locale est beaucoup plus rapide que la mémoire globale, les calculs sont beaucoup plus rapides.
Code du noyau __kernel void ComputeBlockLocal(int offset_n1, int offset_n2, __global const nbcoord_t* mass, __global const nbcoord_t* y, __global nbcoord_t* f, int yoff, int foff, int points_count, int stride) { int n1 = get_global_id(0) + offset_n1; __global const nbcoord_t* rx = y + yoff; __global const nbcoord_t* ry = rx + stride; __global const nbcoord_t* rz = rx + 2 * stride; __global const nbcoord_t* vx = rx + 3 * stride; __global const nbcoord_t* vy = rx + 4 * stride; __global const nbcoord_t* vz = rx + 5 * stride; __global nbcoord_t* frx = f + foff; __global nbcoord_t* fry = frx + stride; __global nbcoord_t* frz = frx + 2 * stride; __global nbcoord_t* fvx = frx + 3 * stride; __global nbcoord_t* fvy = frx + 4 * stride; __global nbcoord_t* fvz = frx + 5 * stride; nbcoord_t x1 = rx[n1]; nbcoord_t y1 = ry[n1]; nbcoord_t z1 = rz[n1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; __local nbcoord_t x2[NBODY_DATA_BLOCK_SIZE]; __local nbcoord_t y2[NBODY_DATA_BLOCK_SIZE]; __local nbcoord_t z2[NBODY_DATA_BLOCK_SIZE]; __local nbcoord_t m2[NBODY_DATA_BLOCK_SIZE];
Cuda
L'implémentation de la plate-forme NVidia CUDA est un peu plus simple qu'OpenCL, nous n'avons pas besoin de créer le contexte de l'appareil et de gérer la file d'attente d'exécution nous-mêmes (au moins jusqu'à ce que nous voulions effectuer une implémentation multi-GPU). Comme dans le cas d'OpenCL, nous devons allouer de la mémoire sur le GPU, y copier nos données, puis nous pouvons démarrer le cœur de calcul.
Vous pouvez en savoir plus sur le travail avec CUDA
ici .
Code de lancement du noyau CUDA dim3 grid(count / block_size); dim3 block(block_size); size_t shared_size(4 * sizeof(nbcoord_t) * block_size); kfcompute <<< grid, block, shared_size >>> (... ...);
Contrairement à OpenCL, CUDA ne spécifie pas la gamme complète des itérations (dans l'implémentation OpenCL c'est global_range), mais définit la taille de la grille et la taille des blocs dans la grille, en conséquence, le calcul du numéro de corps actuel change légèrement, sinon le noyau est très similaire à OpenCL, à l'exception d'autres noms fonctions de synchronisation et de spécification pour la mémoire partagée. Une autre caractéristique distinctive utile de CUDA est que nous pouvons spécifier la taille requise de la mémoire partagée au démarrage du noyau. Comme dans l'implémentation OpenCL, au début de chaque bloc d'itération, nous copions une partie des données dans la mémoire partagée, puis travaillons avec cette mémoire à partir de tous les threads du bloc.
Code du noyau CUDA __global__ void kfcompute(int offset_n2, const nbcoord_t* y, int yoff, nbcoord_t* f, int foff, const nbcoord_t* mass, int points_count, int stride) { int n1 = blockDim.x * blockIdx.x + threadIdx.x; const nbcoord_t* rx = y + yoff; const nbcoord_t* ry = rx + stride; const nbcoord_t* rz = rx + 2 * stride; nbcoord_t x1 = rx[n1]; nbcoord_t y1 = ry[n1]; nbcoord_t z1 = rz[n1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; extern __shared__ nbcoord_t shared_xyzm_buf[]; nbcoord_t* x2 = shared_xyzm_buf; nbcoord_t* y2 = x2 + blockDim.x; nbcoord_t* z2 = y2 + blockDim.x; nbcoord_t* m2 = z2 + blockDim.x; for(int b2 = 0; b2 < points_count; b2 += blockDim.x) { int n2 = b2 + offset_n2 + threadIdx.x;
Tableau 3. La dépendance de la fonction de temps de calcul (en secondes) fn sur le nombre de corps en interaction N pour diverses implémentations GPU et de meilleures implémentations CPU| N | 4096 | 8192 | 16384 | 32768 | 65536 | 131072 |
|---|
| openmp + bloc + optimisation | 0,0128 | 0,0495 | 0,194 | 0,774 | --- | --- |
| OpenCL + demi NVidia K80 | 0,004 | 0,008 | 0,026 | 0,134 | 0,322 | 1.18 |
| CUDA + demi NVidia K80 | 0,004 | 0,008 | 0,0245 | 0,115 | 0,291 | 1.13 |
Où obtenir NVidia K80Les implémentations OpenCL et CUDA ont été lancées sur le service gratuit
Colab de Google, qui donne accès aux ordinateurs NVidia K80. Vous pouvez en savoir plus sur l'utilisation de ce service sur le
concentrateur En général, le résultat est décevant, seulement 5-6 fois plus rapide que la mise en œuvre du CPU. Même si nous effectuons des calculs sur l'ensemble du K80, nous obtiendrons une accélération jusqu'à 12 fois, mais puisque Étant donné que la complexité de la tâche est quadratique, nous pourrons traiter dans un délai raisonnable non pas 32768 corps en interaction, mais 131072, ce qui n'est que 4 fois plus.
Approximation des fonctions fn
Si vous regardez de près la fonction, qui est définie par la force d'attraction de deux corps, il est clair qu'elle diminue de façon quadratique avec la distance. Par conséquent, nous pouvons calculer avec précision la force d'interaction entre des corps proches et approximativement entre des corps éloignés. Une approche célèbre
est un algorithme de
treecode proposé par D. Barnes et P. Hat. Un
arbre d'octree est construit dans l'espace simulé, contenant dans ses feuilles les coordonnées et les masses des corps simulés. Les nœuds parents contiennent le centre de masse, la masse totale des nœuds enfants et le rayon de la sphère décrit autour des corps des nœuds enfants. La racine de l'arbre contient le centre de masse de tous les corps, leur masse totale et le rayon de la sphère décrite autour d'eux. Lors du calcul de la force d'interaction, la distance à la racine de l'arbre est d'abord prise en compte si le rapport de la distance au nœud sur son rayon est supérieur à une constante
lambdacrit , alors la racine est considérée comme un corps avec des coordonnées égales aux coordonnées du centre de masse des corps qui y sont inclus, et une masse égale à la somme des masses des nœuds filles, si le rapport est inférieur ou égal à
lambdacrit , puis la procédure est répétée récursivement pour chaque nœud enfant. Si une feuille d'un arbre est atteinte, alors la force d'interaction est considérée de la manière habituelle. Ainsi, si un corps est très éloigné du groupe compact d'autres corps, ce groupe lui est présenté comme un seul corps, et la force d'interaction est calculée non pas jusqu'à chaque corps, mais seulement jusqu'à un corps. Pour cette raison, la complexité de l'algorithme diminue avec
O(N2) avant
O(N log(N)) au prix d'une perte de précision.
Dans mon approche, j'ai décidé de ne pas utiliser l'
arbre octree , mais l'
arbre kd, car il est plus facile à utiliser et a une surcharge de stockage inférieure à l'octree.
Revenons à l'implémentation sur le CPU. Un nœud kd-tree peut être représenté sous la forme d'une classe contenant des pointeurs vers les descendants gauche et droit et des informations sur les coordonnées et la masse:
Noeud d'arbre Kd class node { node* m_left;
Avec cette méthode de stockage de l'arbre, nous avons deux options possibles pour parcourir l'arbre: soit utiliser la récursivité explicite, soit utiliser la pile nous-mêmes. J'ai opté pour la deuxième option.
Calcul de la force d'interaction en traversant un arbre nbvertex_t force_compute(const nbvertex_t& v1, const nbcoord_t mass1) { nbvertex_t total_force; node* stack_data[MAX_STACK_SIZE] = {}; node** stack = stack_data; node** stack_head = stack; *stack++ = m_root; while(stack != stack_head) { node* curr = *--stack; const nbcoord_t distance_sqr((v1 - curr->m_mass_center).norm()); if(distance_sqr > curr->m_radius_sqr) {
Comme dans le cas de l'implémentation CPU «exacte», la fonction de calcul de force est appelée pour chaque corps. Le cycle à travers tous les corps peut être facilement parallélisé en utilisant les directives OpenMP.
Mais les itérations de boucles voisines dans ce cas feront référence à des parties complètement différentes de l'arborescence, ce qui ne permet pas une utilisation efficace du cache du processeur. Pour surmonter ce problème, tous les corps doivent être traversés non pas dans l'ordre d'origine, mais dans l'ordre dans lequel les corps sont situés dans les feuilles de l'arbre kd, puis des itérations voisines se produiront pour les corps qui sont proches dans l'espace, et traverseront l'arbre selon presque les mêmes chemins.
Traversée des feuilles des arbres template<class Visitor> void traverse(Visitor visit) { node* stack_data[MAX_STACK_SIZE] = {}; node** stack = stack_data; node** stack_head = stack; *stack++ = m_root; while(stack != stack_head) { node* curr = *--stack; if(curr->m_radius_sqr > 0) {
Cette implémentation a un autre problème - il n'existe aucun moyen universel de paralléliser une telle traversée d'arbre. Mais nous pouvons complètement changer la façon dont l'arbre est stocké en mémoire - nous pouvons stocker tous les nœuds dans un tableau linéaire et abandonner complètement le stockage des pointeurs vers les descendants, par analogie avec la construction d'un
tas binaire . Lors du démarrage de la numérotation des nœuds avec
1 si le nœud est dans le numéro de cellule
i , puis son enfant gauche est dans la cellule
2i , bon enfant dans la cellule

et le parent dans la cellule
i/2 . Le nœud droit correspondant à la gauche avec le numéro
i aura un certain nombre
i+1 . Le tableau lui-même aura une longueur
2N , et tous les nœuds feuilles seront situés dans le dernier
N en outre, des nœuds étroitement espacés seront situés dans des cellules proches du réseau. La fonction de traversée de l'arbre changera un peu, mais le cadre reste le même:
Calcul de la force en traversant un arbre dans un tableau nbvertex_t force_compute(const nbvertex_t& v1, const nbcoord_t mass1) { nbvertex_t total_force; size_t stack_data[MAX_STACK_SIZE] = {}; size_t* stack = stack_data; size_t* stack_head = stack; *stack++ = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { size_t curr = *--stack; const nbcoord_t distance_sqr((v1 - m_mass_center[curr]).norm()); if(distance_sqr > m_radius_sqr[curr]) { total_force += force(v1, m_mass_center[curr], mass1, m_mass[curr]); } else { size_t left(left_idx(curr)); size_t rght(rght_idx(curr)); if(rght < m_body_n.size()) { *stack++ = rght; } if(left < m_body_n.size()) { *stack++ = left; } } } return total_force; }
Mais ce ne sont pas toutes les possibilités que le stockage des nœuds dans le tableau nous ouvre - nous pouvons refuser la pile lors du déplacement. Pour ce faire, dans la branche de code dans laquelle nous allons vers les enfants du nœud, nous ajoutons la fonction pour calculer le nœud suivant (
nextup ), et dans la branche dans laquelle nous calculons la force d'interaction, nous ajoutons le calcul du nœud suivant avec le sous-arbre actuel ignoré (
skipdown )
Pour ignorer le sous-arbre actuel, nous devons descendre à la racine (direction
D ), alors que nous sommes dans le descendant droit, dès que nous atteignons la gauche, nous allons au sous-arbre droit correspondant (direction
R ), si nous arrivons à la racine, alors la traversée de l'arbre est terminée.
Code de fonction de saut de sous-arborescence index_t skip_down(index_t idx) {
 Figure 8. Ignorer un sous-arbre skipdown .
Figure 8. Ignorer un sous-arbre skipdown .Pour aller au nœud suivant, si possible, allez vers l'enfant gauche (direction
U ), et s'il n'y a pas de descendant, passez au nœud suivant «d'en bas» en utilisant la fonction
skipdown .
Aller au code de fonction de nœud suivant index_t next_up(index_t idx, index_t tree_size) { index_t left = left_idx(idx); if(left < tree_size) { return left; } return skip_down(idx); }
 Figure 9. Transitions vers le nœud suivant nextup .
Figure 9. Transitions vers le nœud suivant nextup .Il pourrait sembler que nous avons remplacé la récursivité par une boucle
t a n d i s q u e en fonction
s k i p d o w n , et un tel remplacement ne donne rien, mais voyons comment déterminer si un nœud avec un nombre
je descendant droit. Cela peut être fait simplement en vérifiant son nombre impair (le bon enfant est dans la cellule
), pour cela il suffit de calculer
i \ & 1 . C'est-à-dire on divise le nombre
i deux si le bit le moins significatif est mis à un. Mais cela peut être fait sans boucle, dans de nombreux processeurs, il existe une instruction
find first set qui renvoie la position du premier bit défini, en l'utilisant, nous transformons la boucle en quatre instructions de processeur.
Code de fonction de sous-arborescence optimisé index_t skip_down(index_t idx) { idx = idx >> (__builtin_ffs(~idx) - 1); return left2right(idx); }
Après cela, nous pouvons exclure la pile de la fonction de traversée de l'arbre et la remplacer par une paire
skipdown+nextup , après cela, la fonction semble encore plus simple:
Calcul de la force en traversant un arbre dans un tableau sans utiliser de pile nbvertex_t force_compute(const nbvertex_t& v1, const nbcoord_t mass1) const { nbvertex_t total_force; size_t curr = NBODY_HEAP_ROOT_INDEX; size_t tree_size = m_mass_center.size(); do { const nbcoord_t distance_sqr((v1 - m_mass_center[curr]).norm()); if(distance_sqr > m_radius_sqr[curr]) { total_force += force(v1, m_mass_center[curr], mass1, m_mass[curr]); curr = skip_down(curr); } else { curr = next_up(curr, tree_size); } } while(curr != NBODY_HEAP_ROOT_INDEX); return total_force; }
Au total, nous avons obtenu six combinaisons de traversée d'arbre et de calcul de force. Comparez ces approches en termes de temps et de qualité de calcul. Nous prenons comme mesure de la qualité la variation relative de l'énergie totale du système après 100 itérations. En tant que système modèle, nous prenons deux «galaxies» en interaction consistant en16384corps chacun.Tableau 4. Combinaisons de la méthode de traversée des arbres et du calcul de la force| Traversée d'arbre / type de calcul de force | Arbre avec pile | «Pile» avec une pile | «Tas» sans pile |
|---|
| Itérations par numéro de corps | cycle + arbre | cycle + tas | cycle + tas |
|---|
| Contournement des feuilles | arbre imbriqué + arbre | nestedtree + heap | nestedtree + heapstackless |
|---|

| 
|
| a) Dépendance temporelle du calcul de fonction fn du rapport de la distance critique au nœud d'arbre à son rayon ( λcrit ) | b) La dépendance du changement relatif d'énergie dans le système du rapport de la distance critique au nœud d'arbre à son rayon ( λcrit ) |
| Figure 10. Résultats du calcul de fonction fn de différentes manières sur le CPU (nombre de corps N=32768 ) |
On peut voir que la mise en œuvre de 'nestedtree + tree' est désespérément en retard de vitesse, car il manque de simultanéité. Les implémentations avec l'emplacement des nœuds d'arborescence dans le tableau et l'indexation comme dans un segment binaire sont en tête. Le changement relatif d'énergie est négligeable pour toutes les variantes avecλcrit>1 .
Également sur la fig. 10a montre que pour (λcrit<10 ) toutes les variantes de calcul de fonction fn dépasser de manière significative en vitesse la version la plus optimisée du calcul exact («openmp + bloc + optimisation»), avec une augmentation supplémentaire λcrit les implémentations avec un arbre perdent la version exacte.Traversée de l'arbre GPU
J'ai essayé de contourner l'arborescence du GPU en utilisant à la fois la technologie OpenCL et CUDA. L'option de stockage des nœuds sous la forme d'un arbre a été immédiatement rejetée, et seules les options ont été laissées avec le stockage de l'arbre dans un tableau avec indexation comme dans un tas binaire. En général, l'implémentation du cœur de calcul n'est pas très différente de la version CPU.Noyau OpenCL pour calculer la force en traversant un arbre (traversée dans l'ordre des corps de numérotation) __kernel void ComputeTreeBH(int offset_n1, int points_count, int tree_size, __global const nbcoord_t* y, __global nbcoord_t* f, __global const nbcoord_t* tree_cmx, __global const nbcoord_t* tree_cmy, __global const nbcoord_t* tree_cmz, __global const nbcoord_t* tree_mass, __global const nbcoord_t* tree_crit_r2) { int n1 = get_global_id(0) + offset_n1; int stride = points_count; __global const nbcoord_t* rx = y; __global const nbcoord_t* ry = rx + stride; __global const nbcoord_t* rz = rx + 2 * stride; __global const nbcoord_t* vx = rx + 3 * stride; __global const nbcoord_t* vy = rx + 4 * stride; __global const nbcoord_t* vz = rx + 5 * stride; __global nbcoord_t* frx = f; __global nbcoord_t* fry = frx + stride; __global nbcoord_t* frz = frx + 2 * stride; __global nbcoord_t* fvx = frx + 3 * stride; __global nbcoord_t* fvy = frx + 4 * stride; __global nbcoord_t* fvz = frx + 5 * stride; nbcoord_t x1 = rx[n1]; nbcoord_t y1 = ry[n1]; nbcoord_t z1 = rz[n1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int stack_data[MAX_STACK_SIZE] = {}; int stack = 0; int stack_head = stack; stack_data[stack++] = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { int curr = stack_data[--stack]; nbcoord_t dx = x1 - tree_cmx[curr]; nbcoord_t dy = y1 - tree_cmy[curr]; nbcoord_t dz = z1 - tree_cmz[curr]; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > tree_crit_r2[curr]) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tree_mass[curr] / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; } else { int left = left_idx(curr); int rght = rght_idx(curr); if(left < tree_size) { stack_data[stack++] = left; } if(rght < tree_size) { stack_data[stack++] = rght; } } } frx[n1] = vx[n1]; fry[n1] = vy[n1]; frz[n1] = vz[n1]; fvx[n1] = res_x; fvy[n1] = res_y; fvz[n1] = res_z; }
Dans la première version, la traversée de l'arborescence commençait dans l'ordre de numérotation des corps de la matrice d'origine, de sorte que les threads voisins contournaient des parties complètement différentes de l'arborescence, ce qui affectait négativement les performances du cache de mémoire GPU. Par conséquent, dans le deuxième mode de réalisation, une traversée a été appliquée, à partir du haut de l'arborescence, dans ce cas, les threads voisins commencent à traverser l'arbre le long du même chemin, car les cimes des arbres voisins sont à proximité et dans l'espace. Il est également important que nous choisissions la numérotation dans le tableau des nœuds d'arbre non pas à partir de zéro, mais dans l'un, dans ce cas, les feuilles de l'arbre sont stockées dans la seconde moitié du tableau, et avec un nombre de corps égal à la puissance de deux, nous aurons un accès égal à la mémoire par l'index tn1.Noyau OpenCL pour calculer la force en traversant un arbre (traversant les nœuds de numérotation d'un arbre) __kernel void ComputeHeapBH(int offset_n1, int points_count, int tree_size, __global const nbcoord_t* y, __global nbcoord_t* f, __global const nbcoord_t* tree_cmx, __global const nbcoord_t* tree_cmy, __global const nbcoord_t* tree_cmz, __global const nbcoord_t* tree_mass, __global const nbcoord_t* tree_crit_r2, __global const int* body_n) { int tree_offset = points_count - 1 + NBODY_HEAP_ROOT_INDEX; int stride = points_count; int tn1 = get_global_id(0) + offset_n1 + tree_offset; int n1 = body_n[tn1]; nbcoord_t x1 = tree_cmx[tn1]; nbcoord_t y1 = tree_cmy[tn1]; nbcoord_t z1 = tree_cmz[tn1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int stack_data[MAX_STACK_SIZE] = {}; int stack = 0; int stack_head = stack; stack_data[stack++] = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { int curr = stack_data[--stack]; nbcoord_t dx = x1 - tree_cmx[curr]; nbcoord_t dy = y1 - tree_cmy[curr]; nbcoord_t dz = z1 - tree_cmz[curr]; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > tree_crit_r2[curr]) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tree_mass[curr] / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; } else { int left = left_idx(curr); int rght = rght_idx(curr); if(left < tree_size) { stack_data[stack++] = left; } if(rght < tree_size) { stack_data[stack++] = rght; } } } __global const nbcoord_t* vx = y + 3 * stride; __global const nbcoord_t* vy = y + 4 * stride; __global const nbcoord_t* vz = y + 5 * stride; __global nbcoord_t* frx = f; __global nbcoord_t* fry = frx + stride; __global nbcoord_t* frz = frx + 2 * stride; __global nbcoord_t* fvx = frx + 3 * stride; __global nbcoord_t* fvy = frx + 4 * stride; __global nbcoord_t* fvz = frx + 5 * stride; frx[n1] = vx[n1]; fry[n1] = vy[n1]; frz[n1] = vz[n1]; fvx[n1] = res_x; fvy[n1] = res_y; fvz[n1] = res_z; }
En parcourant l'ordre de numérotation des nœuds d'arbre, nous avons obtenu une augmentation des performances. Mais cette option peut également être améliorée. La mémoire globale dans laquelle les nœuds d'arbre sont actuellement situés est optimisée pour un accès partagé , c'est-à-dire les threads d'un groupe doivent lire les mots situés dans un bloc de mémoire. Dans notre cas, la traversée de l'arborescence commence le long des mêmes chemins, et nous demandons les mêmes données avec tous les threads du groupe, mais plus nous allons plus loin dans l'arborescence, les chemins des threads voisins divergent de plus en plus, et nous devons demander des données différentes, ce qui réduit plusieurs fois les performances du sous-système mémoire. Mais les nœuds de chaque sous-arbre appartenant au même niveau sont situés dans des cellules mémoire relativement proches. C'est-à-dire
lors de la traversée du reste de l'arborescence, les threads voisins du cœur de calcul n'accèdent pas aux mêmes noeuds de l'arborescence, mais proches de la mémoire. Pour optimiser cet accès mémoire, une mémoire de texture peut être utilisée. Mais il y a un hic. Pour le moment, les textures ne prennent pas en charge le travail avec des données de double précision (nous voulons calculer avec précision). Mais dans CUDA, il existe une fonction __hiloint2double qui collecte un nombre double précision à partir de deux entiers.Code de demande de nombre à double précision à partir d'une texture contenant des entiers template<> struct nb1Dfetch<double> { typedef double4 vec4; static __device__ double fetch(cudaTextureObject_t tex, int i) { int2 p(tex1Dfetch<int2>(tex, i)); return __hiloint2double(py, px); } static __device__ vec4 fetch4(cudaTextureObject_t tex, int i) { int ii(2 * i); int4 p1(tex1Dfetch<int4>(tex, ii)); int4 p2(tex1Dfetch<int4>(tex, ii + 1)); vec4 d4 = {__hiloint2double(p1.y, p1.x), __hiloint2double(p1.w, p1.z), __hiloint2double(p2.y, p2.x), __hiloint2double(p2.w, p2.z) }; return d4; } };
Dans ce cas, deux implémentations ont été effectuées, dans un chaque élément de l'arborescence (x, y, z, tree_crit_r2) a été demandé indépendamment, et dans la deuxième implémentation, ces demandes ont été combinées. La demande de masse du nœud se produit beaucoup moins fréquemment, uniquement si la condition r2> tree_crit_r2 [curr] est remplie , il est donc inutile de combiner cette demande avec les autres. Une autre caractéristique utile du cadre CUDA est la possibilité de contrôler le rapport entre les tailles du cache L1 et la taille de la mémoire partagée ( cudaFuncSetCacheConfig ). En cas de traversée d'arbre, nous n'utilisons pas de mémoire partagée, nous pouvons donc augmenter le cache L1 au détriment de celui-ci.Noyau CUDA pour calculer la force en traversant un arbre (en parcourant l'ordre de numérotation des nœuds d'arbre) __global__ void kfcompute_heap_bh_tex(int offset_n1, int points_count, int tree_size, nbcoord_t* f, cudaTextureObject_t tree_xyzr, cudaTextureObject_t tree_mass, const int* body_n) { nb1Dfetch<nbcoord_t> tex; int tree_offset = points_count - 1 + NBODY_HEAP_ROOT_INDEX; int stride = points_count; int tn1 = blockDim.x * blockIdx.x + threadIdx.x + offset_n1 + tree_offset; int n1 = body_n[tn1]; nbvec4_t xyzr = tex.fetch4(tree_xyzr, tn1); nbcoord_t x1 = xyzr.x; nbcoord_t y1 = xyzr.y; nbcoord_t z1 = xyzr.z; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int stack_data[MAX_STACK_SIZE] = {}; int stack = 0; int stack_head = stack; stack_data[stack++] = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { int curr = stack_data[--stack]; nbvec4_t xyzr2 = tex.fetch4(tree_xyzr, curr); nbcoord_t dx = x1 - xyzr2.x; nbcoord_t dy = y1 - xyzr2.y; nbcoord_t dz = z1 - xyzr2.z; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > xyzr2.w) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tex.fetch(tree_mass, curr) / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; } else { int left = nbody_heap_func<int>::left_idx(curr); int rght = nbody_heap_func<int>::rght_idx(curr); if(left < tree_size) { stack_data[stack++] = left; } if(rght < tree_size) { stack_data[stack++] = rght; } } } f[n1 + 3 * stride] = res_x; f[n1 + 4 * stride] = res_y; f[n1 + 5 * stride] = res_z; }
Une analyse du programme dans le profileur nvprof a montré que même lors de l'utilisation de la mémoire de texture pour stocker un arbre, il y a toujours une charge très élevée sur la mémoire globale.En effet, dans CUDA, toute la mémoire du noyau qui est adressée à des adresses `` calculées '' est stockée dans la mémoire globale, et en conséquence la pile nécessaire pour parcourir l'arborescence est située dans la mémoire globale et ronge une partie importante de la bande passante de la puce mémoire, car la pile a chaque thread d'exécution, et il y a beaucoup de threads.Mais, heureusement, nous savons déjà traverser un arbre sans utiliser de pile. En complément du noyau de calcul précédent avec les fonctions de calcul du prochain nœud d'arbre, nous obtenons un nouveau noyau, de plus, plus compact.CUDA core pour calculer la force en traversant un arbre sans utiliser de pile __global__ void kfcompute_heap_bh_stackless(int offset_n1, int points_count, int tree_size, nbcoord_t* f, cudaTextureObject_t tree_xyzr, cudaTextureObject_t tree_mass, const int* body_n) { nb1Dfetch<nbcoord_t> tex; int tree_offset = points_count - 1 + NBODY_HEAP_ROOT_INDEX; int stride = points_count; int tn1 = blockDim.x * blockIdx.x + threadIdx.x + offset_n1 + tree_offset; int n1 = body_n[tn1]; nbvec4_t xyzr = tex.fetch4(tree_xyzr, tn1); nbcoord_t x1 = xyzr.x; nbcoord_t y1 = xyzr.y; nbcoord_t z1 = xyzr.z; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int curr = NBODY_HEAP_ROOT_INDEX; do { nbvec4_t xyzr2 = tex.fetch4(tree_xyzr, curr); nbcoord_t dx = x1 - xyzr2.x; nbcoord_t dy = y1 - xyzr2.y; nbcoord_t dz = z1 - xyzr2.z; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > xyzr2.w) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tex.fetch(tree_mass, curr) / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; curr = nbody_heap_func<int>::skip_idx(curr); } else { curr = nbody_heap_func<int>::next_up(curr, tree_size); } } while(curr != NBODY_HEAP_ROOT_INDEX); f[n1 + 3 * stride] = res_x; f[n1 + 4 * stride] = res_y; f[n1 + 5 * stride] = res_z; }
Les performances des cœurs exécutés sur le GPU dépendent fortement de la taille des blocs dans lesquels nous divisons la tâche. Dépend de cette taille le nombre de registres, de mémoire locale et d'autres ressources disponibles pour chaque thread de calcul. Vous devez également garder à l'esprit qu'en attendant l'accès à la mémoire dans un thread, un autre thread peut effectuer des calculs sur le processeur de shader, ainsi, avec un nombre suffisant de threads s'exécutant simultanément sur un processeur, le temps d'accès à la mémoire sera caché derrière les calculs. Par conséquent, avant de comparer les performances de nos cœurs, nous devons calculer la taille de bloc optimale pour chacun d'eux. Faisons une comparaison sur la moitié disponible de NVidia K80.Tableau 5. La dépendance de la fonction de temps de calcul (en secondes) fn GPU N=131072 λcrit=10| / | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 1024 |
|---|
| opencl+dense | 5.77 | 2.84 | 1.46 | 1.13 | 1.15 | 1.14 | 1.14 | 1.13 |
| cuda+dense | 5.44 | 2.55 | 1.27 | 0.96 | 0.97 | 0.99 | 0.99 | - |
| opencl+heap+cycle | 5.88 | 5.65 | 5.74 | 5.96 | 5.37 | 5.38 | 5.35 | 5.38 |
| opencl+heap+nested | 4.54 | 3,68 | 3.98 | 5.25 | 5.46 | 5.41 | 5.48 | 5.31 |
| cuda+heap+nested | 3,62 | 2,81 | 2,68 | 4.26 | 4.84 | 4.75 | 4.8 | 4.67 |
| cuda+heap+nested+tex | 2.6 | 1.51 | 0.912 | 0.7 | 1.85 | 1.75 | 1.69 | 1.61 |
| cuda+heap+nested+tex+stackless | 2.3 | 1.29 | 0.773 | 0.5 | 0.51 | 0.52 | 0.52 | 0.52 |
6. ( ) fn GPU N=1M λcrit=4| / | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 1024 |
|---|
| opencl+dense | 366 | 179 | 89.9 | 69.3 | 70.3 | 69.1 | 68.9 | 68.0 |
| cuda+dense | 346 | 162 | 79.6 | 60.8 | 60.8 | 60.7 | 59.6 | - |
| opencl+heap+cycle | 16.2 | 18.2 | 20,1 | 21.2 | 21.2 | 21.3 | 21.2 | 21.1 |
| opencl+heap+nested | 10.5 | 7.63 | 6.38 | 8.23 | 9.95 | 9.89 | 9.65 | 9.66 |
| cuda+heap+nested | 8.67 | 6.44 | 5.39 | 5.93 | 8.65 | 8.61 | 8.41 | 8.27 |
| cuda+heap+nested+tex | 6.38 | 3,57 | 2.13 | 1.44 | 3.56 | 3.46 | 3.30 | 3.29 |
| cuda+heap+nested+tex+stackless | 5.78 | 3.19 | 1.83 | 1.21 | 1.11 | 1.10 | 1.11 | 1.13 |
Une situation difficile, mais, contrairement à la version CPU du parcours de l'arborescence, il est clair que chaque étape d'optimisation apporte des résultats tangibles. La mise en œuvre de «opencl + heap + cycle» est presque 6 fois plus lente qu'une solution exacte avec calcul de fonction complètefn .
Une implémentation de 'opencl + heap + nested', dans laquelle une traversée d'arbre commence à partir de nœuds voisins, 1,4 fois plus rapide que la précédente, car La mémoire cache est mieux utilisée. Dans l'implémentation de 'cuda + heap + nested', le cache L1 a été augmenté au détriment de la mémoire partagée, ce qui a augmenté la vitesse de 1,4 fois, bien qu'il soit possible que dans l'implémentation de cuda, le noyau de compilation soit compilé de manière plus optimale (dans les versions de 'opencl + dense' et 'cuda + les cœurs denses sont identiques et les performances de la version de cuda sont environ 1,2 fois supérieures). L'augmentation la plus importante de la vitesse de calcul (3,8 fois) est obtenue lorsque l'arbre est situé dans la mémoire de texture et que les requêtes sur les éléments du nœud d'arbre sont combinées. L'implémentation avec traversée d'arbre sans utiliser la pile 'cuda + heap + nested + tex + stackless' est également 1,4 fois plus rapide.dans ce document, toute la bande passante du bus mémoire est utilisée uniquement pour accéder aux données sur les nœuds d'arbre et n'est pas dépensée sur la pile. Ainsi, avecλcrit=10 Il a été possible d'obtenir une accélération deux fois par rapport au calcul complet de la fonction fn . Mais
λcrit=10 une valeur excessivement grande du paramètre, sur le graphique de la variation relative de l'énergie dans le système du rapport de la distance critique au nœud d'arbre à son rayon pour la mise en œuvre du CPU, on peut voir que des valeurs inférieures peuvent être utilisées λcritsans perte visible de précision. Essayons de varierλcrit avec les tailles de blocs optimales que nous avons déterminées à l'étape précédente.
| 
|
a) N=128K
| b) N=1M
|
Fig. 11. Dépendance du temps de calcul de la fonction. fn du rapport de la distance critique au nœud d'arbre à son rayon ( λcrit ) pour diverses implémentations d'arborescence GPU
|
On peut voir que pour les petits λcrit toutes les méthodes de calcul de fonction fnaller aux valeurs proches, déterminées par le temps de construction de l'arborescence kd et de préparation des données pour le GPU. De plus, le temps de construction d'un arbre contribue de manière significative au temps totalλcrit≤4, alors cette fois peut être négligée. Il est intéressant de noter que lorsqueN=128K les performances s'améliorent à nouveau en atteignant λcrit=1024cela est probablement dû au fait que tous les threads du GPU tournent autour des mêmes sommets d'arbre en même temps, ce qui améliore l'utilisation du cache L1 et élimine complètement la `` divergence de branche '' . On voit également que la meilleure performance pour une implémentation sans pile (cuda + heap + nested + tex + stackless), elle surpasse la version avec la pile à environ1.4−1.5fois. D'autres implémentations sont plusieurs fois plus lentes. Pour consolider le résultat, nous allons calculer le temps sur un GPU avec une architecture plus récente.Résultats de lancement sur GeForce GTX 1080 Ti (précision unique)7. ( ) fn GPU N=1M λcrit=4| / | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 1024 |
|---|
| opencl+dense | 47.8 | 23.4 | 11.6 | 11.59 | 12.8 | 12.8 | 12.8 | 12.8 |
| cuda+dense | 49.0 | 23.8 | 11.9 | 11.8 | 11.7 | 11.7 | 11.7 | 11.7 |
| opencl+heap+cycle | 7.48 | 8.26 | 7.73 | 7.36 | 7.33 | 7.27 | 7.25 | 7.26 |
| opencl+heap+nested | 1.33 | 1.20 | 1.41 | 2.46 | 2.53 | 2.49 | 2.44 | 2.47 |
| cuda+heap+nested | 1.23 | 1.10 | 1.31 | 2.28 | 2.29 | 2.28 | 2.23 | 2.25 |
| cuda+heap+nested+tex | 0.88 | 0.68 | 0.654 | 1.6 | 1.6 | 1.6 | 1.6 | 1.6 |
| cuda+heap+nested+tex+stackless | 0.71 | 0.47 | 0.45 | 0.43 | 0.43 | 0.42 | 0.41 | 0.395 |

|
12. fn ( λcrit ) GPU
|
Lorsque vous utilisez la GeForce GTX 1080 Ti pour le calcul, la différence entre les marches d'arbre avec une pile et sans pile est jusqu'à deux fois, à condition de négliger le temps nécessaire à la construction de l'arbre. Ce fait nous pousse à garantir qu'à l'avenir le portage vers le GPU et la construction d'un arbre. Une comparaison des tableaux 5-7 montre qu'il n'y a pas de taille de bloc optimale unique pour un nombre différent de corps, et encore plus pour différentes architectures GPU, de plus, la différence de temps de calcul peut atteindre plusieurs fois, même si vous ne tenez pas compte des valeurs limites. Ainsi, avant de longs calculs, il est logique de déterminer la taille de bloc optimale pour chaque configuration.La principale chose que nous avons obtenue est la capacité de modéliser l'interaction par paire d'un peu plus d'un million de corps (220) pendant un temps raisonnable sur un, pas le dernier GPU. Sur les nouveaux GPU (Tesla V100), apparemment, il sera possible de traiter déjà environ deux millions de corps en interaction dans un délai raisonnable, car leurs performances sont environ quatre fois supérieures à la moitié de la Tesla K80. Bien que ce nombre soit également incomparable avec le nombre d'étoiles même dans les galaxies naines, comme le Petit Nuage de Magellan , mais, à mon avis, est impressionnant.Conclusion
L'utilisation de méthodes intégrées pour résoudre des équations différentielles permet de résoudre des problèmes similaires avec un haut niveau de précision à un coût relativement faible, et l'application d'algorithmes pour approximer la fonction de calcul des forces d'attraction par paire nous permet de traiter des volumes colossaux de corps en interaction. Ainsi, contrairement aux extraterrestres des «tâches à trois corps», nous sommes en mesure de résoudre le problèmeN corps, et pour un petit nombre de corps et pour des amas d'étoiles entiers, mais au prix d'une perte de précision.Visualisation
Pour ceux qui ont lu jusqu'à la fin, je donnerai quelques vidéos supplémentaires avec visualisation de l'évolution des systèmes de corps.Simulation d'une collision de deux galaxies. Le nombre total de corps est de 60 mille.Modélisation de l'évolution d'une galaxie composée d'un million d'étoiles. Au centre se trouve un corps d'une masse de 99% du total. Les particules simples sont presque indiscernables. Déjà plus comme une vague dans une goutte de liquide. Colorisé selon le module de vitesse. Basse vitesse - bleu, moyen - vert, haut - rouge. On voit qu'au centre la vitesse est plus élevée, et diminue progressivement jusqu'aux bords, et la plus basse dans le plan équatorial.Une simulation plus «dynamique» de l'évolution d'une galaxie d'un million d'étoiles. Au centre se trouve un corps d'une masse de 10% du total. Le corps central affecte beaucoup moins le reste. D'abord, les «étoiles» s'envolent, puis se regroupent en plusieurs grappes, et à la fin forment à nouveau une grande grappe.En cours de modélisation, environ 5% des «étoiles» ont quitté irrévocablement la région initiale.À la 10e seconde, elle ressemble beaucoup à la galaxie de roue de charrette existante .Le code du projet peut être trouvé sur le github .