Et si vous avez une idée d'une protéine fraîche et saine, et que vous voulez l'obtenir en réalité? Par exemple, aimeriez-vous créer un vaccin contre
H. pylori (comme l'
équipe slovène à iGEM 2008 ) en créant une protéine hybride qui combine des fragments de flagelline d'
E. Coli qui stimulent la réponse immunitaire avec la flagelline
H. pylori habituelle?
H. pylori Hybrid Flagellin Design Présenté par l'équipe slovène à iGEM 2008Étonnamment, nous sommes très près de créer toutes les protéines que nous voulons sans quitter le cahier de Jupyter, grâce aux derniers développements en génomique, en biologie synthétique et plus récemment dans les laboratoires de cloud computing.
Dans cet article, je vais montrer le code Python de l'idée d'une protéine à son expression dans une cellule bactérienne, sans toucher à une pipette ni parler à personne. Le coût total ne sera que de quelques centaines de dollars! En utilisant la
terminologie de Vijaya Pande de A16Z , il s'agit de Biologie 2.0.
Plus précisément, dans l'article, le code Python du laboratoire cloud effectue les opérations suivantes:
- Synthèse d' une séquence d'ADN qui code pour n'importe quelle protéine que je veux.
- Clonage de cet ADN synthétique dans un vecteur pouvant l'exprimer.
- Transformation des bactéries avec ce vecteur et confirmation de l'expression.
Configuration de Python
Tout d'abord, les paramètres généraux de Python qui sont nécessaires pour tout bloc-notes Jupyter. Nous importons des modules Python utiles et créons des fonctions utilitaires, principalement pour la visualisation des données.
Codeimport re import json import logging import requests import itertools import numpy as np import seaborn as sns import pandas as pd import matplotlib as mpl import matplotlib.pyplot as plt from io import StringIO from pprint import pprint from Bio.Seq import Seq from Bio.Alphabet import generic_dna from IPython.display import display, Image, HTML, SVG def uprint(astr): print(astr + "\n" + "-"*len(astr)) def show_html(astr): return display(HTML('{}'.format(astr))) def show_svg(astr, w=1000, h=1000): SVG_HEAD = '''<?xml version="1.0" standalone="no"?><!DOCTYPE svg PUBLIC "-//W3C//DTD SVG 1.1//EN" "http://www.w3.org/Graphics/SVG/1.1/DTD/svg11.dtd">''' SVG_START = '''<svg viewBox="0 0 {w:} {h:}" version="1.1" xmlns="http://www.w3.org/2000/svg" xmlns:xlink= "http://www.w3.org/1999/xlink">''' return display(SVG(SVG_HEAD + SVG_START.format(w=w, h=h) + astr + '</svg>')) def table_print(rows, header=True): html = ["<table>"] html_row = "</td><td>".join(k for k in rows[0]) html.append("<tr style='font-weight:{}'><td>{}</td></tr>".format('bold' if header is True else 'normal', html_row)) for row in rows[1:]: html_row = "</td><td>".join(row) html.append("<tr style='font-family:monospace;'><td>{:}</td></tr>".format(html_row)) html.append("</table>") show_html(''.join(html)) def clean_seq(dna): dna = re.sub("\s","",dna) assert all(nt in "ACGTN" for nt in dna) return Seq(dna, generic_dna) def clean_aas(aas): aas = re.sub("\s","",aas) assert all(aa in "ACDEFGHIKLMNPQRSTVWY*" for aa in aas) return aas def Images(images, header=None, width="100%"):
Laboratoires cloud
Comme AWS ou tout autre cloud informatique, le laboratoire de cloud possède des équipements de biologie moléculaire, ainsi que des robots qu'il loue sur Internet. Vous pouvez envoyer des instructions à vos robots en cliquant sur quelques boutons de l'interface ou en écrivant du code qui les programme vous-même. Il n'est pas nécessaire d'écrire vos propres protocoles, comme je le ferai ici, une partie importante de la biologie moléculaire est des tâches de routine standard, il est donc généralement préférable de s'appuyer sur un protocole étranger fiable qui a montré une bonne interaction avec les robots.
Récemment, un certain nombre d'entreprises dotées de laboratoires cloud sont apparues:
Transcriptic ,
Autodesk Wet Lab Accelerator (bêta et construit sur la base de Transcriptic),
Arcturus BioCloud (bêta),
Emerald Cloud Lab (bêta),
Synthego (n'a pas encore commencé). Il existe même des entreprises construites au-dessus de laboratoires cloud tels que
Desktop Genetics , spécialisé dans CRISPR.
Des articles scientifiques sur l'utilisation des laboratoires cloud dans la vraie science commencent à paraître.
Au moment d'écrire ces lignes, seul Transcriptic est dans le domaine public, nous allons donc l'utiliser. Si je comprends bien, la plupart des activités de transcription reposent sur l'automatisation de protocoles courants, et l'écriture de vos propres protocoles en Python (comme je le ferai dans cet article) est moins courante.
 «Cellule de travail» transcriptique avec réfrigérateurs en bas et divers équipements de laboratoire sur le stand
«Cellule de travail» transcriptique avec réfrigérateurs en bas et divers équipements de laboratoire sur le standJe donnerai aux robots Transcriptic des instructions sur l'
auto-protocole . Autoprotocol est un langage basé sur JSON pour écrire des protocoles pour les robots de laboratoire (et les humains, pour ainsi dire). Le protocole automatique est principalement réalisé sur
cette bibliothèque Python . Le langage a été créé à l'origine et est toujours pris en charge par Transcriptic, mais, si je comprends bien, il est complètement ouvert. Il y a une bonne
documentation .
Une idée intéressante est que vous pouvez écrire des instructions pour les personnes dans des laboratoires distants, par exemple en Chine ou en Inde, sur l'auto-protocole et potentiellement obtenir des avantages en utilisant à la fois des personnes (leur jugement) et des robots (manque de jugement). Nous devons mentionner
ici protocoles.io , il s'agit d'une tentative de normalisation des protocoles pour améliorer la reproductibilité, mais pour les humains, pas pour les robots.
"instructions": [ { "to": [ { "well": "water/0", "volume": "500.0:microliter" } ], "op": "provision", "resource_id": "rs17gmh5wafm5p" }, ... ]
Exemple de fragment autoprotocoleParamètres Python pour la biologie moléculaire
En plus d'importer des bibliothèques standard, j'aurai besoin de certains utilitaires biologiques moléculaires spécifiques. Ce code est principalement destiné aux protocoles automatiques et transcriptiques.
Le concept de «volume mort» se retrouve souvent dans le code. Cela signifie la dernière goutte de liquide que les robots Transcriptic ne peuvent pas prendre avec une pipette des tubes (car ils ne peuvent pas la voir!). Vous devez passer beaucoup de temps pour vous assurer que les flacons contiennent suffisamment de matière.
Code import autoprotocol from autoprotocol import Unit from autoprotocol.container import Container from autoprotocol.protocol import Protocol from autoprotocol.protocol import Ref
Synthèse d'ADN et biologie synthétique
Malgré sa connexion avec la biologie synthétique moderne, la synthèse d'ADN est une technologie assez ancienne. Pendant des décennies, nous avons pu fabriquer des oligonucléotides (c'est-à-dire des séquences d'ADN jusqu'à 200 bases). Cependant, c'était toujours cher, et la chimie n'a jamais permis de longues séquences d'ADN. Récemment, il est devenu possible à un prix raisonnable de synthétiser des gènes entiers (jusqu'à des milliers de bases). Cette réalisation ouvre véritablement l'ère de la «biologie synthétique».
La génomique synthétique de Craig Venter
a poussé la biologie synthétique le plus loin en
synthétisant un organisme entier - plus d'un million de bases de long. À mesure que la longueur de l'ADN augmente, le problème n'est plus la synthèse, mais l'assemblage (c'est-à-dire l'assemblage de séquences d'ADN synthétisées). Avec chaque assemblage, vous pouvez doubler la longueur d'ADN (ou plus), donc après une dizaine d'itérations, vous obtenez une
molécule assez longue ! La distinction entre synthèse et assemblage devrait bientôt devenir claire pour l'utilisateur final.
La loi de Moore?
Le prix de la synthèse d'ADN baisse assez rapidement, passant de plus de 0,30 $ il y a deux ans à environ 0,10 $ aujourd'hui, mais elle se développe plus comme des bactéries que comme des processeurs. En revanche, les prix du séquençage de l'ADN baissent plus rapidement que la loi de Moore. Un objectif de
0,02 $ par base est défini comme un point d'inflexion où vous pouvez remplacer de nombreuses manipulations d'ADN chronophages par une simple synthèse. Par exemple, à ce prix, vous pouvez synthétiser un plasmide entier de 3 ko pour
60 $ et sauter un tas de biologie moléculaire. J'espère que nous y parviendrons dans quelques années.
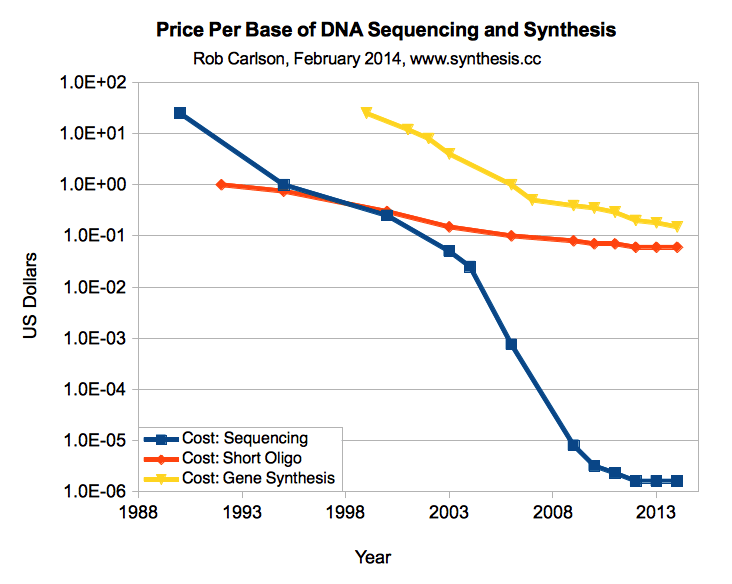 Prix de synthèse d'ADN comparés aux prix de séquençage d'ADN, prix pour 1 base (Carlson, 2014)
Prix de synthèse d'ADN comparés aux prix de séquençage d'ADN, prix pour 1 base (Carlson, 2014)Entreprises de synthèse d'ADN
Il existe plusieurs grandes entreprises dans le domaine de la synthèse d'ADN: IDT est le plus grand producteur d'oligonucléotides et peut également produire des «fragments de gènes» (
gBlocks ) plus longs (jusqu'à 2
kb ).
Gen9 ,
Twist et
DNA 2.0 se spécialisent généralement dans les séquences d'ADN plus longues - ce sont des sociétés de synthèse de gènes. Il existe également de nouvelles sociétés intéressantes, telles que
Cambrian Genomics et
Genesis DNA , qui travaillent sur des méthodes de synthèse de nouvelle génération.
D'autres sociétés, comme
Amyris ,
Zymergen et
Ginkgo Bioworks , utilisent l'ADN synthétisé par ces sociétés pour travailler au niveau du corps.
La génomique synthétique le fait aussi, mais elle synthétise l'ADN lui-même.
Ginkgo a récemment
conclu un accord avec Twist pour faire 100 millions de bases: le plus gros accord que j'ai vu. Cela prouve que nous vivons dans le futur, Twist a même annoncé un code promotionnel sur Twitter: lorsque vous achetez 10 millions de bases d'ADN (presque tout le génome de la levure!), Vous obtenez 10 millions supplémentaires gratuitement.
 Offre Twitter Twist Niche
Offre Twitter Twist NichePremière partie: Conception de l'expérience
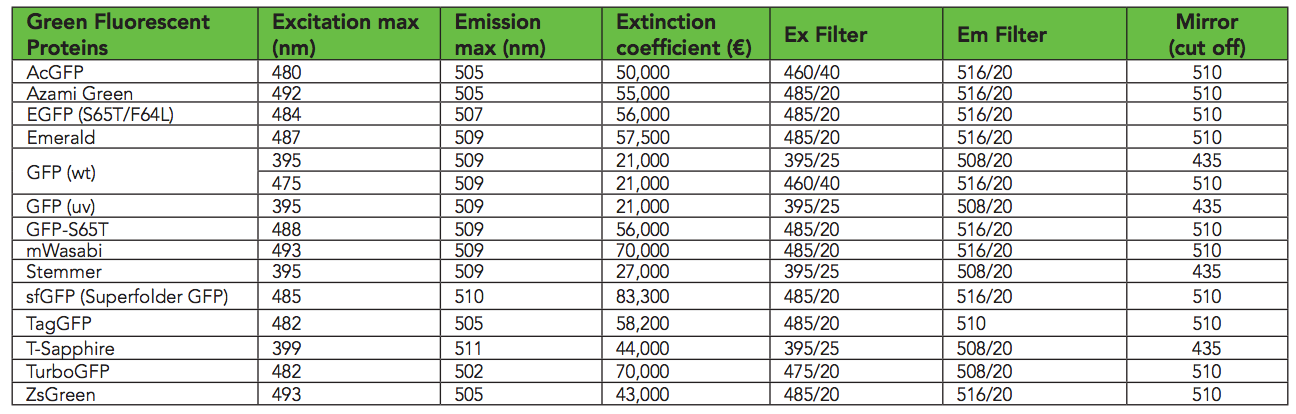
Protéine fluorescente verte
Dans cette expérience, nous synthétisons une séquence d'ADN pour une simple
protéine fluorescente verte (GFP). La protéine GFP a été découverte pour la première fois dans une
méduse fluorescente sous lumière ultraviolette. Il s'agit d'une protéine extrêmement utile car il est facile de détecter son expression simplement en mesurant la fluorescence. Il existe des options GFP qui produisent du jaune, du rouge, de l'orange et d'autres couleurs.
Il est intéressant de voir comment diverses mutations affectent la couleur d'une protéine, et c'est un problème d'apprentissage automatique potentiellement intéressant. Plus récemment, il faudrait passer beaucoup de temps en laboratoire pour cela, mais maintenant je vais vous montrer que c'est (presque) aussi simple que d'éditer un fichier texte!
Techniquement, mon GFP est une option Super Folder (sfGFP) avec quelques mutations pour améliorer la qualité.
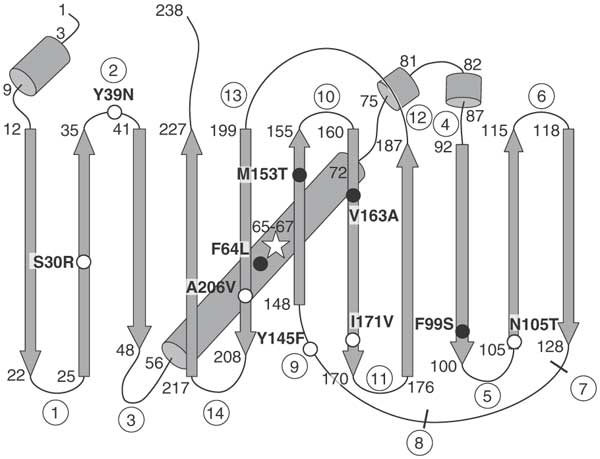 Dans superfolder-GFP (sfGFP), certaines mutations lui confèrent certaines propriétés utiles.
Dans superfolder-GFP (sfGFP), certaines mutations lui confèrent certaines propriétés utiles.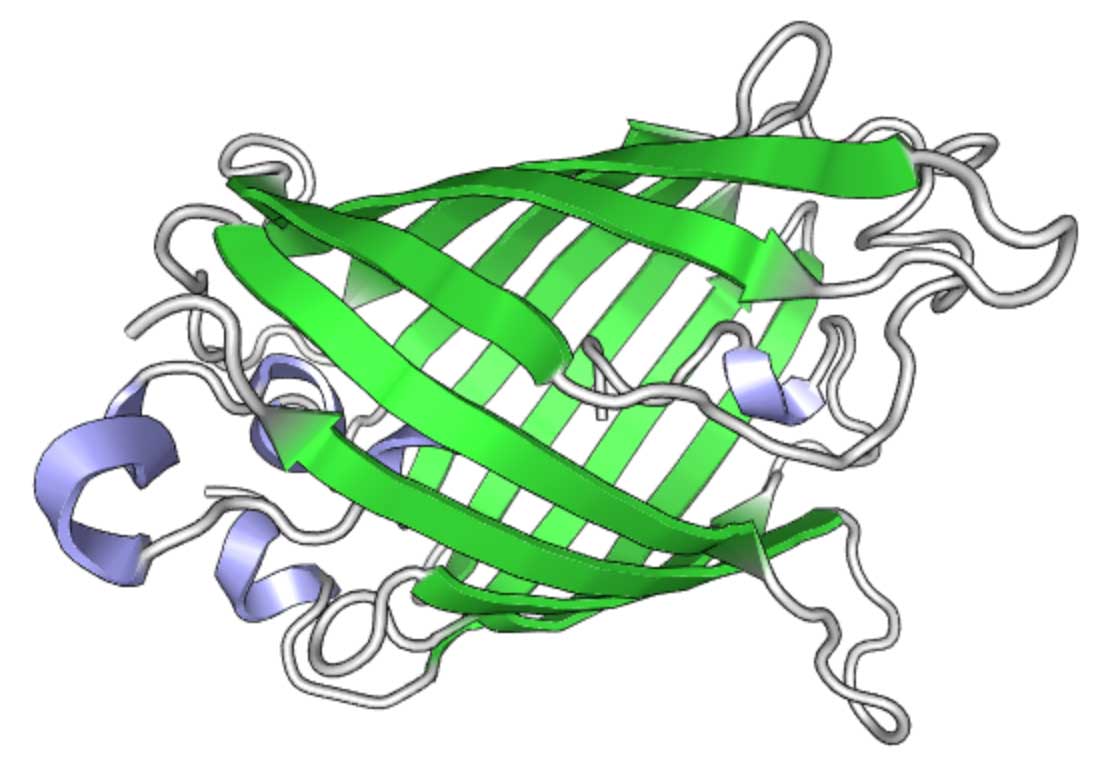 Structure GFP (visualisée à l'aide de PV )
Structure GFP (visualisée à l'aide de PV )Synthèse de GFP dans Twist
J'ai eu la chance de participer au programme de test alpha de Twist, j'ai donc utilisé leur service de synthèse d'ADN (ils ont gentiment passé ma petite commande - merci, Twist!). Il s'agit d'une nouvelle entreprise dans notre domaine, avec un nouveau processus de synthèse simplifié. Leurs prix sont d'environ
0,10 $ par base ou moins , mais ils sont
toujours en version bêta , et le programme alpha auquel j'ai participé a été fermé. Twist a recueilli environ 150 millions de dollars, de sorte que leur technologie est vivante.
J'ai envoyé ma séquence d'ADN à Twist sous forme de feuille de calcul Excel (il n'y a pas encore d'API, mais je suppose que ce sera bientôt), et ils ont envoyé l'ADN synthétisé directement dans ma boîte dans le laboratoire de transcription (j'ai également utilisé IDT pour la synthèse, mais ils n'ont pas envoyé ADN en transcriptic, ce qui gâche un peu le plaisir).
De toute évidence, ce processus n'est pas encore devenu un cas d'utilisation typique et nécessite un certain support, mais il a fonctionné, de sorte que l'ensemble du pipeline reste virtuel. Sans cela, j'aurais probablement besoin d'avoir accès au laboratoire - de nombreuses entreprises n'enverront pas d'ADN ou de réactifs à leur domicile.
 GFP est inoffensif, donc tout type est mis en évidence
GFP est inoffensif, donc tout type est mis en évidenceVecteur plasmidique
Pour exprimer cette protéine dans des bactéries, le gène doit vivre quelque part, sinon l'ADN synthétique codant pour le gène se dégrade simplement instantanément. En règle générale, en biologie moléculaire, nous utilisons un plasmide, un morceau d'ADN rond qui vit en dehors du génome bactérien et exprime des protéines. Les plasmides sont un moyen pratique pour les bactéries de partager des modules fonctionnels utiles et autonomes, tels que la résistance aux antibiotiques. Il peut y avoir des centaines de plasmides dans une cellule.
La terminologie largement utilisée est qu'un plasmide est un
vecteur et que l'ADN synthétique est une insertion (insertion). Donc, ici, nous essayons de cloner l'insertion dans un vecteur, puis de transformer les bactéries à l'aide du vecteur.
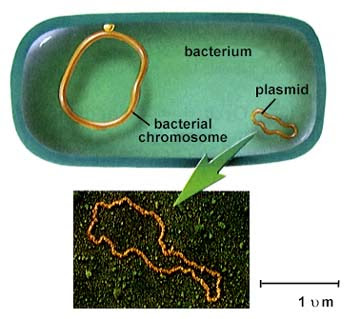 Génome bactérien et plasmide (pas à l'échelle!) ( Wikipedia )
Génome bactérien et plasmide (pas à l'échelle!) ( Wikipedia )pUC19
J'ai choisi un plasmide assez standard dans la série
pUC19 . Ce plasmide est très souvent utilisé, et puisqu'il est disponible dans le cadre de l'inventaire transcriptique standard, nous n'avons pas besoin de leur envoyer quoi que ce soit.
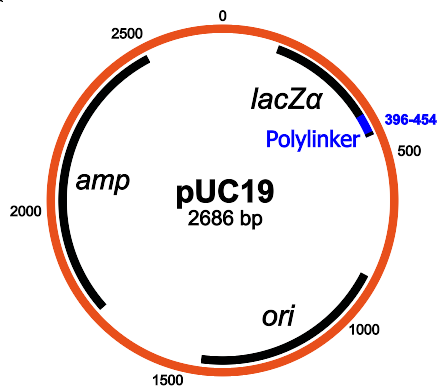 Structure de pUC19: les principaux composants sont le gène de résistance à l'ampicilline, lacZα, MCS / polylinker et l'origine de la réplication (Wikipedia)
Structure de pUC19: les principaux composants sont le gène de résistance à l'ampicilline, lacZα, MCS / polylinker et l'origine de la réplication (Wikipedia)Le PUC19 a une fonction intéressante: puisqu'il contient le gène lacZα, vous pouvez utiliser la méthode de
sélection bleu-blanc dessus et voir dans quelles colonies l'insertion a réussi. Deux produits chimiques sont nécessaires:
IPTG et
X-gal , et le circuit fonctionne comme suit:
- L'IPTG induit l'expression de lacZα.
- Si lacZα est désactivé via l'ADN inséré au site de clonage multiple ( MCS / polylinker ) dans lacZα, alors le plasmide ne peut pas hydrolyser X-gal et ces colonies seront blanches au lieu de bleu.
- Par conséquent, une insertion réussie produit des colonies blanches et une insertion échouée produit des colonies bleues.
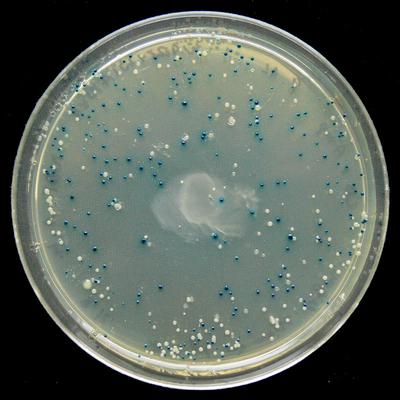 La sélection en bleu et blanc montre où l'expression lacZα a été désactivée ( Wikipedia )La documentation openwetware
La sélection en bleu et blanc montre où l'expression lacZα a été désactivée ( Wikipedia )La documentation openwetware indique:
E. coli DH5α ne nécessite pas d'IPTG pour induire l'expression du promoteur lac, même si un répresseur Lac est exprimé dans la souche. Le nombre de copies de la plupart des plasmides dépasse le nombre de répresseurs dans les cellules. Si vous avez besoin d'une expression maximale, ajoutez IPTG à une concentration finale de 1 mM.
Séquences d'ADN synthétique
Séquence d'ADN SfGFP
Il est facile d'obtenir la séquence d'ADN de sfGFP en prenant
la séquence protéique et en la
codant avec des codons appropriés pour l'organisme hôte (ici,
E. coli ). Il s'agit d'une protéine de taille moyenne avec 236 acides aminés, donc à 10 cents la synthèse d'ADN coûte environ
70 $ par base.
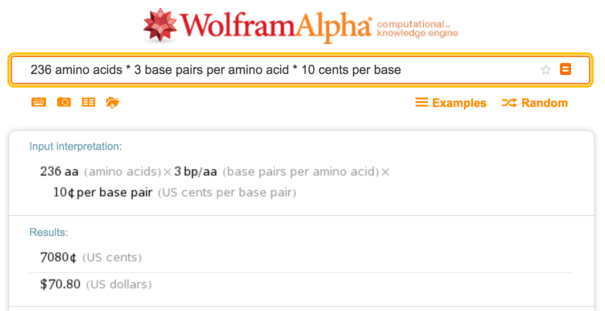 Wolfram Alpha, calcul des coûts de synthèse
Wolfram Alpha, calcul des coûts de synthèseLes 12 premières bases de notre sfGFP sont la
séquence Shine-Delgarno , que j'ai ajoutée moi-même, qui devrait théoriquement augmenter l'expression (AGGAGGACAGCT, puis ATG (
start codon ) démarre la protéine). Selon un outil de calcul développé par
Salis Lab (
diapositives de cours ), on peut s'attendre à une expression moyenne à élevée de notre protéine (taux d'initiation de la traduction de 10 000 «unités arbitraires»).
sfGFP_plus_SD = clean_seq(""" AGGAGGACAGCTATGTCGAAAGGAGAAGAACTGTTTACCGGTGTGGTTCCGATTCTGGTAGAACTGGA TGGGGACGTGAACGGCCATAAATTTAGCGTCCGTGGTGAGGGTGAAGGGGATGCCACAAATGGCAAAC TTACCCTTAAATTCATTTGCACTACCGGCAAGCTGCCGGTCCCTTGGCCGACCTTGGTCACCACACTG ACGTACGGGGTTCAGTGTTTTTCGCGTTATCCAGATCACATGAAACGCCATGACTTCTTCAAAAGCGC CATGCCCGAGGGCTATGTGCAGGAACGTACGATTAGCTTTAAAGATGACGGGACCTACAAAACCCGGG CAGAAGTGAAATTCGAGGGTGATACCCTGGTTAATCGCATTGAACTGAAGGGTATTGATTTCAAGGAA GATGGTAACATTCTCGGTCACAAATTAGAATACAACTTTAACAGTCATAACGTTTATATCACCGCCGA CAAACAGAAAAACGGTATCAAGGCGAATTTCAAAATCCGGCACAACGTGGAGGACGGGAGTGTACAAC TGGCCGACCATTACCAGCAGAACACACCGATCGGCGACGGCCCGGTGCTGCTCCCGGATAATCACTAT TTAAGCACCCAGTCAGTGCTGAGCAAAGATCCGAACGAAAAACGTGACCATATGGTGCTGCTGGAGTT CGTGACCGCCGCGGGCATTACCCATGGAATGGATGAACTGTATAAA""") print("Read in sfGFP plus Shine-Dalgarno: {} bases long".format(len(sfGFP_plus_SD))) sfGFP_aas = clean_aas("""MSKGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLTYG VQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNFNSHNVYITADKQKN GIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHGMDELYK""") assert sfGFP_plus_SD[12:].translate() == sfGFP_aas print("Translation matches protein with accession 532528641")
Lire dans sfGFP plus Shine-Dalgarno: 726 bases de long
La traduction correspond à la protéine avec l'accession 532528641
Séquence d'ADN PUC19
Tout d'abord, je vérifie que la
séquence pUC19 que j'ai téléchargée de l'ONÉ a la bonne longueur et qu'elle inclut le
polylinker attendu.
pUC19_fasta = !cat puc19fsa.txt pUC19_fwd = clean_seq(''.join(pUC19_fasta[1:])) pUC19_rev = pUC19_fwd.reverse_complement() assert all(nt in "ACGT" for nt in pUC19_fwd) assert len(pUC19_fwd) == 2686 pUC19_MCS = clean_seq("GAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTT") print("Read in pUC19: {} bases long".format(len(pUC19_fwd))) assert pUC19_MCS in pUC19_fwd print("Found MCS/polylinker")
Lire en pUC19: 2686 bases de long
Trouvé MCS / polylinker
Nous faisons quelques QC de base pour nous assurer que EcoRI et BamHI ne sont présents dans pUC19 qu'une seule fois (les enzymes de restriction suivantes sont disponibles dans l'inventaire transcriptique par défaut:
PstI ,
PvuII ,
EcoRI ,
BamHI ,
BbsI ,
BsmBI ).
REs = {"EcoRI":"GAATTC", "BamHI":"GGATTC"} for rename, res in REs.items(): assert (pUC19_fwd.find(res) == pUC19_fwd.rfind(res) and pUC19_rev.find(res) == pUC19_rev.rfind(res)) assert (pUC19_fwd.find(res) == -1 or pUC19_rev.find(res) == -1 or pUC19_fwd.find(res) == len(pUC19_fwd) - pUC19_rev.find(res) - len(res)) print("Asserted restriction enzyme sites present only once: {}".format(REs.keys()))
Maintenant, nous regardons la séquence lacZα et vérifions qu'il n'y a rien d'inattendu. Par exemple, il doit commencer par Met et se terminer par un codon stop. Il est également facile de confirmer qu'il s'agit de l'ORF lacZα complet de 324 pb en chargeant la séquence pUC19 dans le
visualiseur Snapgene gratuit.
lacZ = pUC19_rev[2217:2541] print("lacZα sequence:\t{}".format(lacZ)) print("r_MCS sequence:\t{}".format(pUC19_MCS.reverse_complement())) lacZ_p = lacZ.translate() assert lacZ_p[0] == "M" and not "*" in lacZ_p[:-1] and lacZ_p[-1] == "*" assert pUC19_MCS.reverse_complement() in lacZ assert pUC19_MCS.reverse_complement() == pUC19_rev[2234:2291] print("Found MCS once in lacZ sequence")
séquence lacZ: ATGACCATGATTACGCCAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGTAATAGCGAAGAGGCCCGCACCGATCGCCCTTCCCAACAGTTGCGCAGCCTGAATGGCGAATGGCGCCTGATGCGGTATTTTCTCCTTACGCATCTGTGCGGTATTTCACACCGCATATGGTGCACTCTCAGTACAATCTGCTCTGATGCCGCATAG
séquence r_MCS: AAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTC
MCS trouvé une fois dans la séquence lacZ
Assemblage Gibson
L'assemblage d'ADN signifie simplement des fragments de réticulation. Habituellement, vous collectez plusieurs fragments d'ADN dans un segment plus long, puis vous les clonez dans un plasmide ou un génome. Dans cette expérience, je veux cloner un segment d'ADN dans le plasmide pUC19 sous le promoteur lac pour l'expression dans
E. coli .
Il existe de nombreuses méthodes de clonage (par exemple
NEB ,
openwetware ,
addgene ). Ici, je vais utiliser l'assemblage Gibson (
développé par Daniel Gibson dans Synthetic Genomics en 2009), qui n'est pas nécessairement la méthode la moins chère, mais simple et flexible. Il vous suffit de mettre l'ADN que vous souhaitez collecter (avec le chevauchement approprié) dans un tube à essai avec le Gibson Assembly Master Mix, et il s'assemblera!
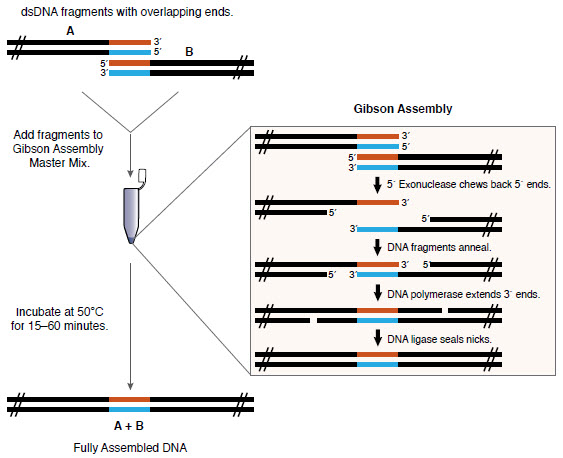 Revue de l'Assemblée Gibson ( ONÉ )
Revue de l'Assemblée Gibson ( ONÉ )Matériel d'origine
On commence avec 100 ng d'ADN synthétique dans 10 µl de liquide. Cela équivaut à 0,21 picomole d'ADN ou à une concentration de 10 ng / μl.
pmol_sfgfp = convert_ug_to_pmol(0.1, len(sfGFP_plus_SD)) print("Insert: 100ng of DNA of length {:4d} equals {:.2f} pmol".format(len(sfGFP_plus_SD), pmol_sfgfp))
Insert: 100ng d'ADN de longueur 726 est égal à 0,21 pmol
Selon
le protocole d'assemblage de
l'ONÉ , c'est assez de matériel source:
L'ONÉ recommande un total de 0,02 à 0,5 picomole de fragments d'ADN lorsque 1 ou 2 fragments sont assemblés dans le vecteur, ou 0,2 à 1,0 picomole de fragments d'ADN lorsque 4 à 6 fragments sont collectés.
0,02-0,5 pmoles * X μl
* L'efficacité de clonage optimisée est de 50 à 100 ng de vecteurs avec 2-3 fois plus d'insertions. Utilisez 5 fois plus d'insertions si la taille est inférieure à 200 bps. Le volume total de fragments de PCR non filtrés dans la réaction d'assemblage de Gibson ne doit pas dépasser 20%.
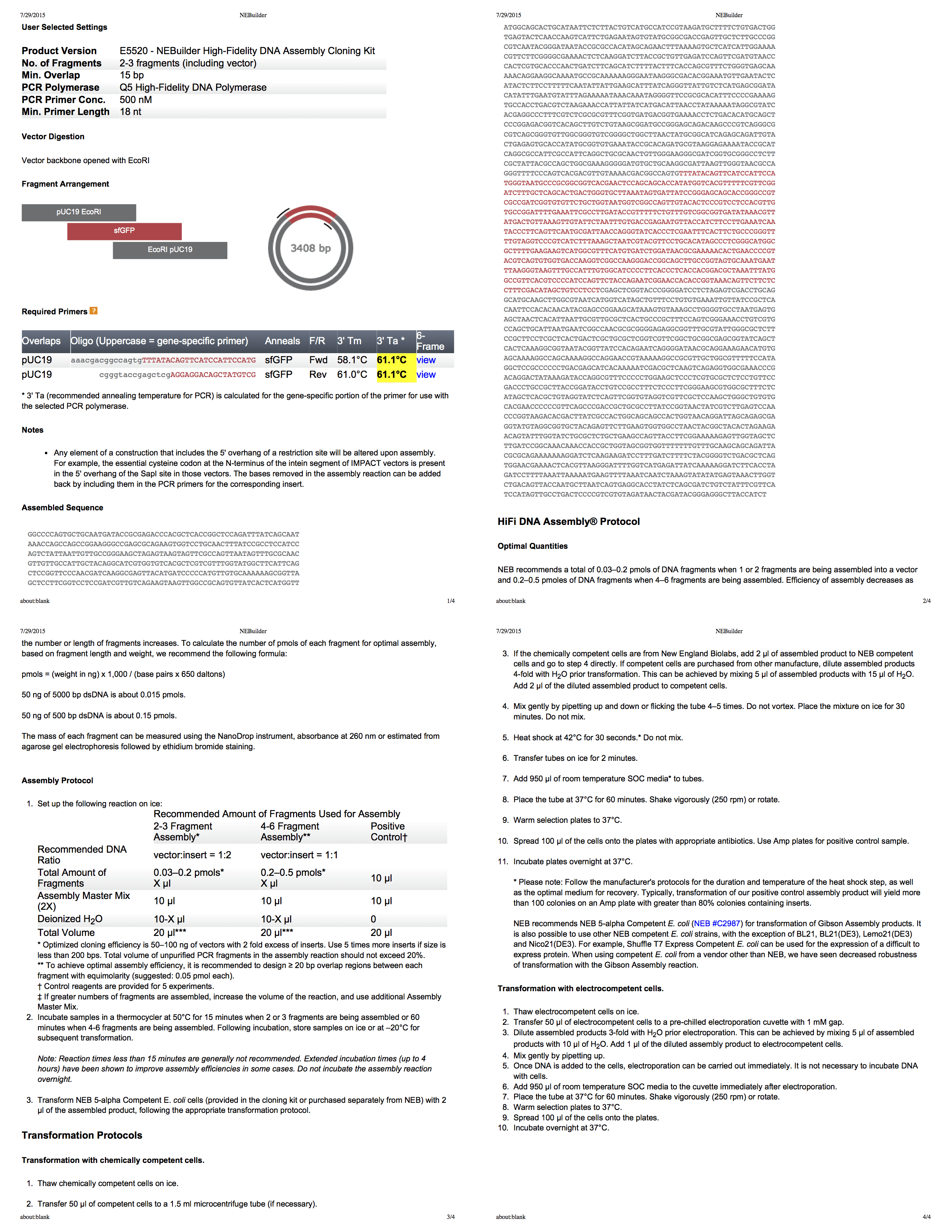
NEBuilder pour l'assemblage Gibson
NEBuilder de Biolab est un très bon outil pour créer un protocole de construction Gibson. Il vous génère même un PDF complet de quatre pages avec toutes les informations. En utilisant cet outil, nous développons un protocole pour couper pUC19 avec EcoRI, puis utilisons la PCR [PCR, la réaction en chaîne par polymérase permet d'obtenir une augmentation significative des petites concentrations de certains fragments d'ADN dans le matériel biologique - env. par.] pour ajouter des fragments de la taille appropriée à l'insertion.
Deuxième partie: expérimenter
L'expérience comprend quatre étapes:
- Réaction d'insertion de chaîne de polymérase pour ajouter du matériel avec une séquence flanquante.
- Couper un plasmide pour permettre l'insertion.
- Assemblage par insertion de Gibson et plasmides.
- Transformation de bactéries à l'aide du plasmide assemblé.
Étape 1. Insertion PCR
L'assemblage Gibson dépend de la séquence d'ADN que vous collectez, ayant une séquence qui se chevauchent (voir le protocole NEB avec les instructions détaillées ci-dessus). En plus d'une simple amplification, la PCR vous permet également d'ajouter une séquence d'ADN flanquante en incluant simplement une séquence supplémentaire dans les amorces (peut également être clonée en
utilisant uniquement OE-PCR ).
Nous synthétisons les amorces selon le protocole NEB ci-dessus. J'ai essayé
le protocole Quickstart sur le site Transcriptic, mais il y a toujours
une commande auto- protocol . Le transcriptic lui-même ne synthétise pas les oligonucléotides, donc après 1-2 jours d'attente, ces amorces apparaissent comme par magie dans mon inventaire (notez que la partie spécifique au gène des amorces est indiquée en majuscule ci-dessous, mais ce ne sont que des choses cosmétiques).
insert_primers = ["aaacgacggccagtgTTTATACAGTTCATCCATTCCATG", "cgggtaccgagctcgAGGAGGACAGCTATGTCG"]
Analyse d'amorce
Vous pouvez analyser les propriétés de ces amorces à l'aide de l'
IDT OligoAnalyzer . PCR
primer dimer , NEB .
Gene-specific portion of flank (uppercase)
Melt temperature: 51C, 53.5C
Full sequence
Melt temperature: 64.5C, 68.5C
Hairpin: -.4dG, -5dG
Self-dimer: -9dG, -16dG
Heterodimer: -6dG
PCR, , PCR. ( ), : . . , — .
Code """ PCR overlap extension of sfGFP according to NEB protocol. v5: Use 3/10ths as much primer as the v4 protocol. v6: more complex touchdown pcr procedure. The Q5 temperature was probably too hot v7: more time at low temperature to allow gene-specific part to anneal v8: correct dNTP concentration, real touchdown """ p = Protocol()
WARNING:root:Low volume for well sfGFP 1 /sfGFP 1 : 2.0:microliter
sfGFP 1 /sfGFP 1 2.0:microliter {'dilution': '0.25ng/ul'}
sfgfp_pcroe_v5_puc19_primer1_10uM 75.0:microliter {}
sfgfp_pcroe_v5_puc19_primer2_10uM 75.0:microliter {}
Consolidated volume 52.0:microliter
Protocol 1. Amplify the insert (oligos previously synthesized)
---------------------------------------------------------------
✓ Protocol analyzed
11 instructions
8 containers
Total Cost: $32.18
Workcell Time: $4.32
Reagents & Consumables: $27.86 : PCR
( )
( ). , , .
D1, E1, F1 2 , 4 8 . (50 ). , .
GelEval , , , , . . GelEval 40 /.
,
, dNTP , , 12,5 , 6 740bp 25 . GelEval 40 x 25 (1 2 ), , .
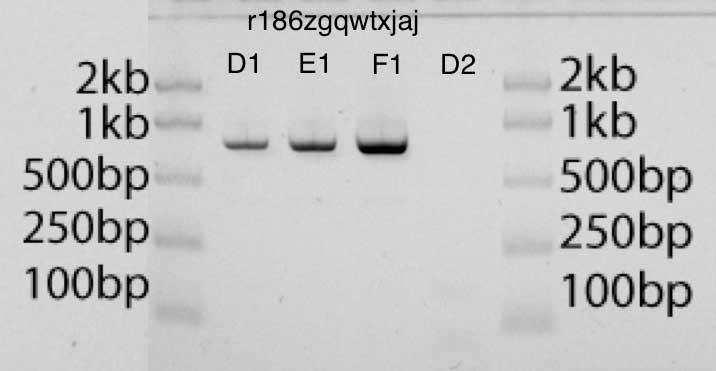 - EcoRI- pUC19, (D1, E1, F1), (D2)
- EcoRI- pUC19, (D1, E1, F1), (D2)PCR
Transcriptic . , .
, . 35 PCR,
PCR . — ! — , PCR , .
 PCR: , 35 42
PCR: , 35 422.
sfGFP pUC19, . NEB,
EcoRI . Transcriptic , :
NEB EcoRI 10x CutSmart ,
NEB pUC19 .
, . , Transcriptic :
Item ID Amount Concentration Price
------------ ------ ------------- ----------------- ------
CutSmart 10x B7204S 5 ml 10 X $19.00
EcoRI R3101L 50,000 units 20,000 units/ml $225.00
pUC19 N3041L 250 µg 1,000 µg/ml $268.00
NEB:
. 10X dH2O 1X. , , , , . 50 5 10x NEBuffer , dH2O.
, 1 λ 1 37°C 50 . , 5-10 10-20 1- .
1 50 .
Code """Protocol for cutting pUC19 with EcoRI.""" p = Protocol() experiment_name = "puc19_ecori_v3" options = {} inv = { 'water': "rs17gmh5wafm5p",
Volumes: re_tube:135.0:microliter water_tube:383.0:microliter EcoRI:30.0:microliter
Consolidated volume: 78.0:microliter
✓ Protocol analyzed
12 instructions
5 containers
Total Cost: $30.72
Workcell Time: $3.38
Reagents & Consumables: $27.34
:
, . .
«» ( 1,5 15 !). , D1 E1 ( ). , EcoRI .
, D1 E1 2,6kb. D2 : , .
Deux photos sur gel sont très différentes. Cela est dû en partie au fait que cette étape Transcriptic n'a pas encore été automatisée.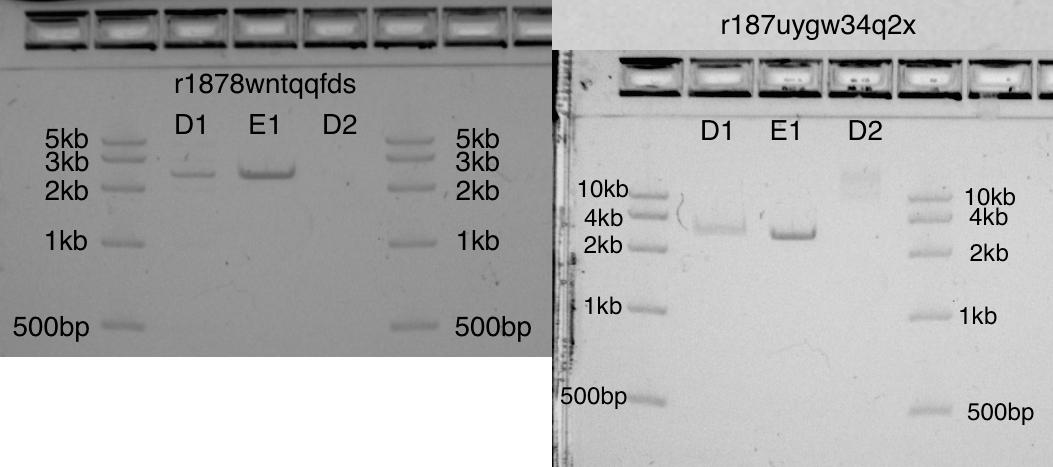 Deux gels montrant pUC19 coupé (2.6kb) dans les bandes D1 et E1, et pUC19 non coupé en D2
Deux gels montrant pUC19 coupé (2.6kb) dans les bandes D1 et E1, et pUC19 non coupé en D2Étape 3. Assemblage Gibson
, — ,
M13 ( )
qPCR , , . , , , .
, M13 , M13.
Code """Debugging transformation protocol: Gibson assembly followed by qPCR and a gel v2: include v3 Gibson assembly""" p = Protocol() options = {} experiment_name = "debug_sfgfp_puc19_gibson_seq_v2" inv = { "water" : "rs17gmh5wafm5p",
WARNING:root:Low volume for well sfgfp_puc19_gibson_v1_clone/sfgfp_puc19_gibson_v1_clone : 11.0:microliter
✓ Protocol analyzed
11 instructions
6 containers
Total Cost: $32.09
Workcell Time: $6.98
Reagents & Consumables: $25.11
: qPCR
qPCR JSON Transcriptic API.
, . API , .
-, :
project_id, run_id = "p16x6gna8f5e9", "r18mj3cz3fku7" api_url = "https://secure.transcriptic.com/hgbrian/{}/runs/{}/data.json".format(project_id, run_id) data_response = requests.get(api_url, headers=tsc_headers) data = data_response.json()
id, «» qPCR:
qpcr_id = data['debug_sfgfp_puc19_gibson_seq_v1_qpcr']['id'] pp_api_url = "https://secure.transcriptic.com/data/{}.json?key=postprocessed_data".format(qpcr_id) data_response = requests.get(pp_api_url, headers=tsc_headers) pp_data = data_response.json()
Ct ( ) . Ct — , . , (, , , ).
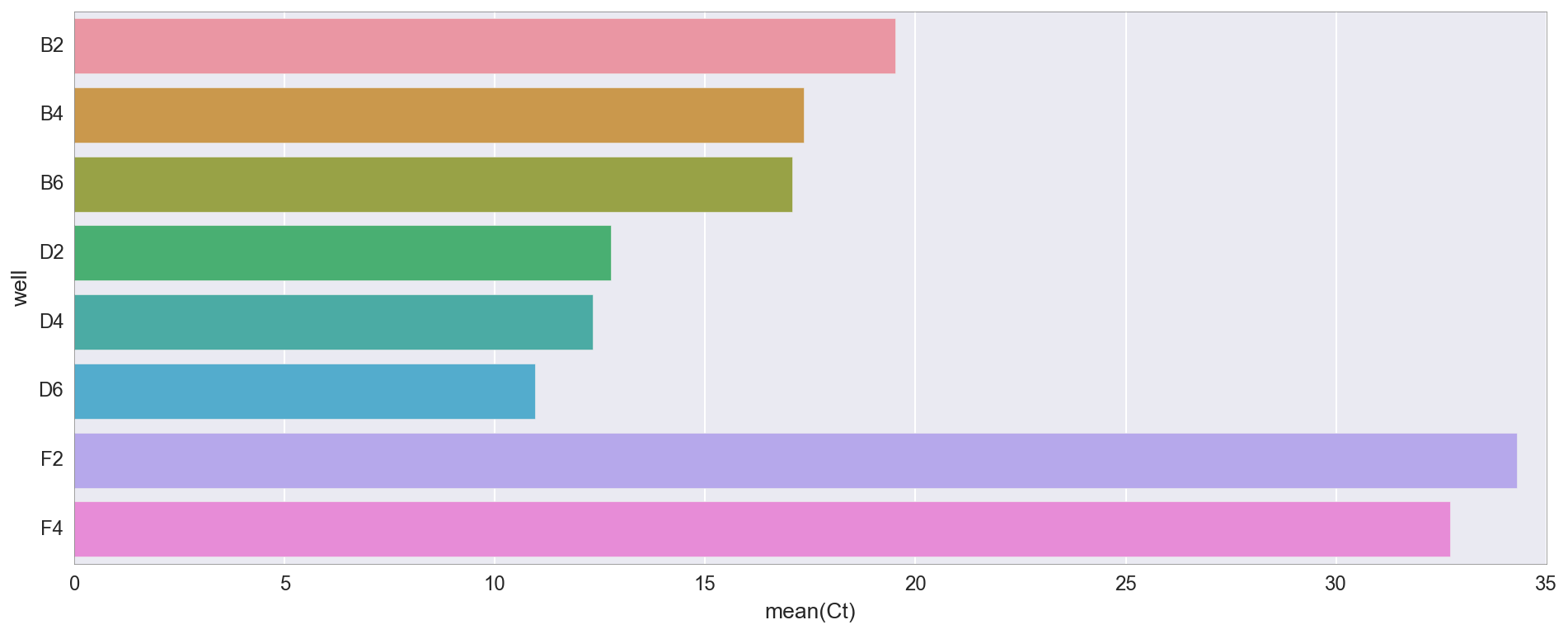
, D2/4/6 ( «v3»), B2/4/6 ( «v1»). v1 v3 , v3 4X NEB, . 30 (F2, F4), -, , .
qPCR, .
f, ax = plt.subplots(figsize=(16,6)) ax.set_color_cycle(['#fb6a4a', '#de2d26', '#a50f15', '#74c476', '#31a354', '#006d2c', '#08519c', '#6baed6']) amp0 = pp_data['amp0']['SYBR']['baseline_subtracted'] _ = [plt.plot(amp0[w_n[well]], label=well) for well in ['B2', 'B4', 'B6', 'D2', 'D4', 'D6', 'F2', 'F4']] _ = ax.set_ylim(0,) _ = plt.title("qPCR (reds=Gibson v1, greens=Gibson v3, blues=control)") _ = plt.legend(bbox_to_anchor=(1, .75), bbox_transform=plt.gcf().transFigure)
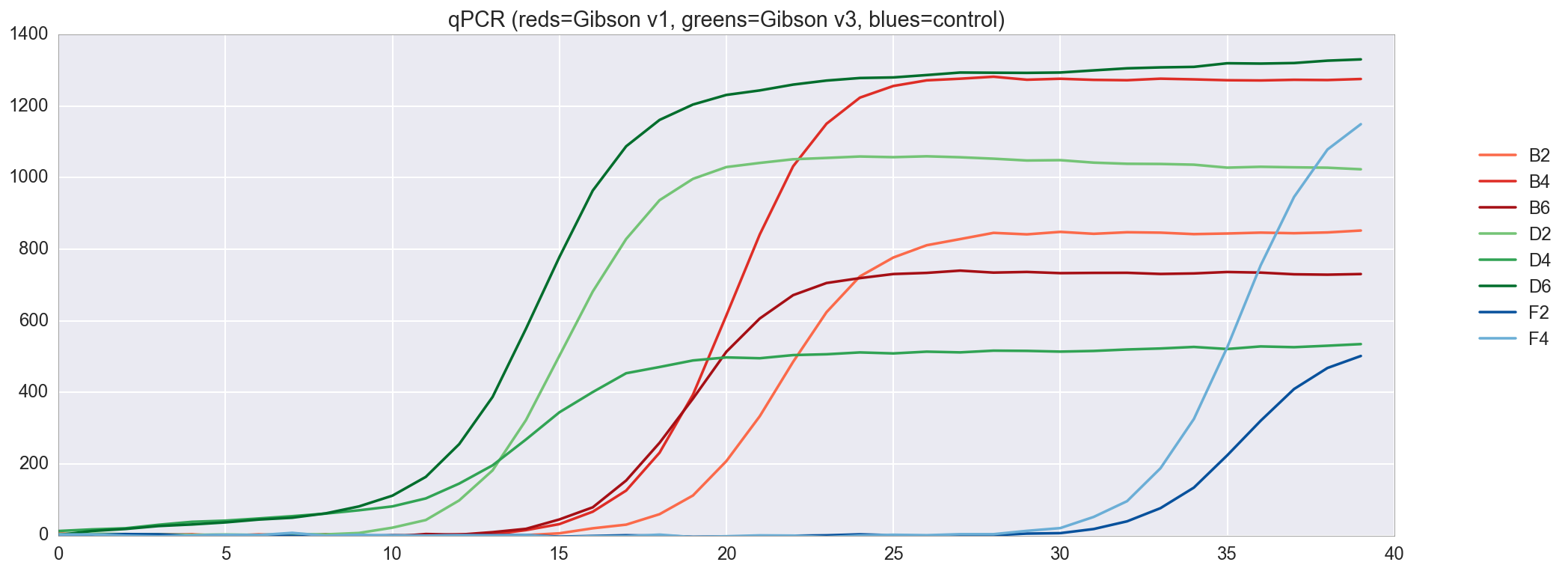
, qPCR , . v3 , v1, .
:
, 1kb B2, B4, B6, D2, D4, D6: ( 740bp, M13 — 40bp ). . , F2 F4 .
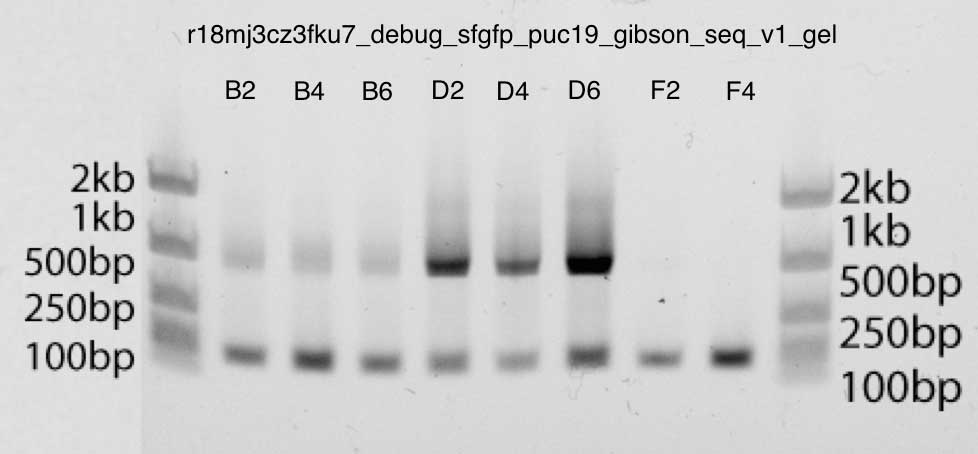 : v3 (D2, D4, D6), qPCR
: v3 (D2, D4, D6), qPCR4.
— .
E. coli sfGFP- pUC19.
Zymo DH5α Mix&Go . — Transcriptic. , , , , . , , .
 Zymo Mix & Go
Zymo Mix & Go— , . (« »), , («, »). , .
. , 37°C. , , , , Transcriptic — , . , , - , . . .
: (, , Mix&Go ); (, ); (, PCR ).
, , , , . , , !
, , , pUC19 (. . sfGFP) . pUC19 , , .
(«6-flat» Transcriptic), , . , , , . .
Code """Simple transformation protocol: transformation with unaltered pUC19""" p = Protocol() experiment_name = "debug_sfgfp_puc19_gibson_v1" inv = { "water" : "rs17gmh5wafm5p",
✓ Protocol analyzed
43 instructions
3 containers
$45.43
:
, ( ) , , . , Transcriptic , .
( ) , . , , , , 55 10 . . , . , , .
( , , ,
E. coli . La croissance est beaucoup plus faible sur les plaques d'ampicilline, bien qu'il y ait beaucoup plus de bactéries, comme prévu).Dans l'ensemble, la transformation a assez bien fonctionné pour se poursuivre, bien qu'il y ait quelques défauts. Plaques de cellules transformées avec pUC19 après 18 heures: sans antibiotique (à gauche) et avec antibiotique (à droite)
Plaques de cellules transformées avec pUC19 après 18 heures: sans antibiotique (à gauche) et avec antibiotique (à droite)Transformation du produit après assemblage
pUC19, , , , sfGFP.
, IPTG X-gal,
- . , pUC19, sfGFP, .
, sfGFP 485 / 510 . , Transcriptic 485/535. , 485 510 . 600 (
OD600 ).
 GFP ( biotek )
GFP ( biotek )IPTG X-gal
IPTG 1M 1:1000. , X-gal 20 / 1:1000 (20 /). , 2000µl LB 2 .
40 X-gal 20 / 40 IPTG 0,1 mM ( 4 IPTG 1M), 30 . , IPTG, X-gal .
Code """Full Gibson assembly and transformation protocol for sfGFP and pUC19 v1: Spread IPTG and X-gal onto plates, then spread cells v2: Mix IPTG, X-gal and cells; spread the mixture v3: exclude X-gal so I can do colony picking better v4: repeat v3 to try other excitation/emission wavelengths""" p = Protocol() options = { "gibson" : False,
Inventory: IPTG/IPTG/IPTG/IPTG/IPTG/IPTG 832.0:microliter {}
Inventory: sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone 57.0:microliter {}
✓ Protocol analyzed
40 instructions
8 containers
Total Cost: $53.20
Workcell Time: $17.35
Reagents & Consumables: $35.86 , «» 96- . (
autopick ).
Code """Pick colonies from plates and grow in amp media and check for fluorescence. v2: try again with a new plate (no blue colonies) v3: repeat with different emission and excitation wavelengths""" p = Protocol() options = {} for k, v in list(options.items()): if v is False: del options[k] experiment_name = "sfgfp_puc19_gibson_pick_v3" def plate_expid(val): """refer to the previous plating experiment's outputs""" plate_exp = "sfgfp_puc19_gibson_plates_v4" return "{}_{}".format(plate_exp, val)
✓ Protocol analyzed
62 instructions
8 containers
Total Cost: $66.38
Workcell Time: $57.59
Reagents & Consumables: $8.78
:
– , , (1-4) (5-6). , , , , IPTG X-gal, Transcriptic.
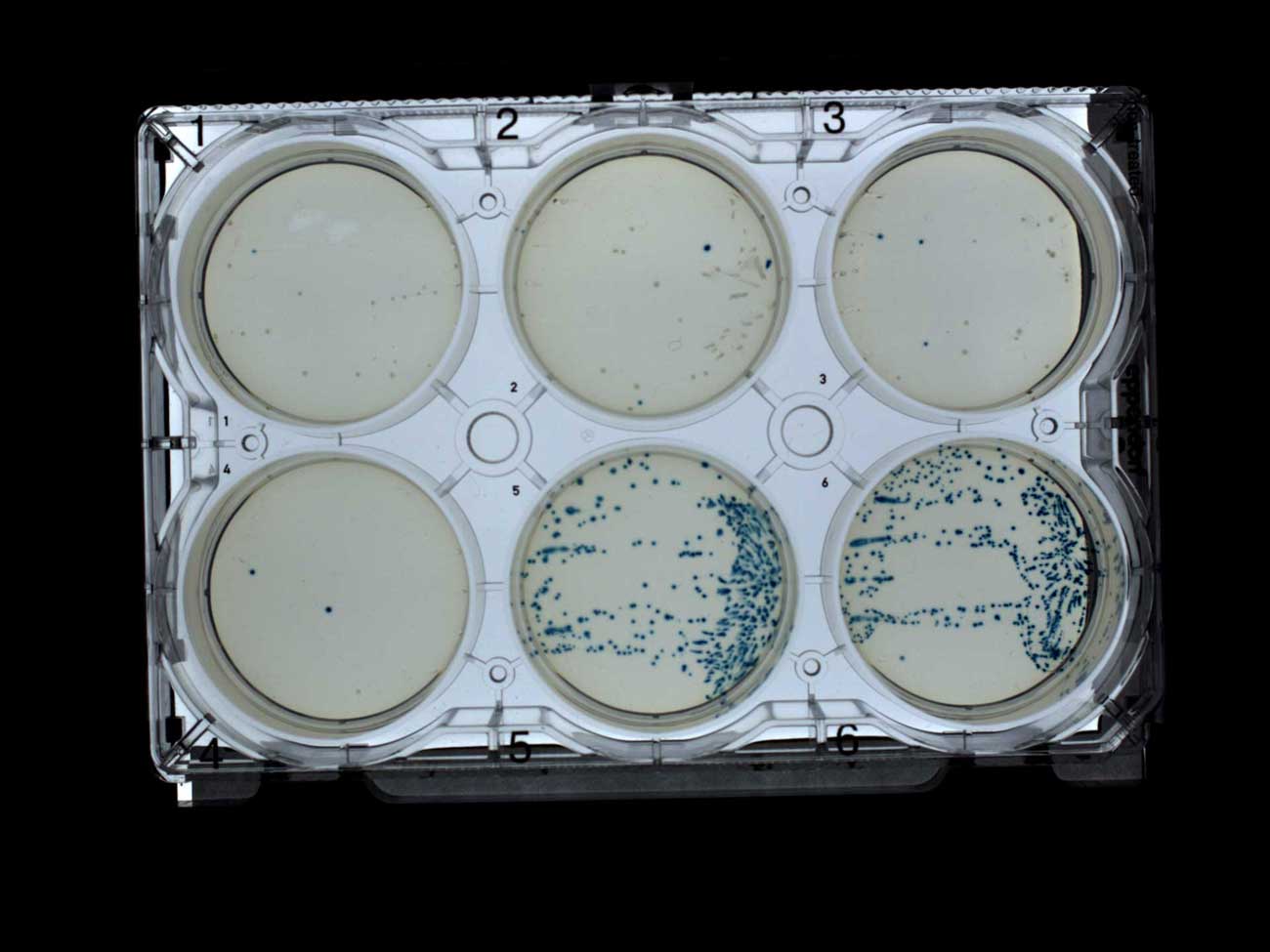 - (1-4) (5-6)
- (1-4) (5-6)- - . (
GraphicsMagick ). , , ( , ).
, Transcriptic. , 10 . , , . . , , , , .
 - (1-4) (5-6),
- (1-4) (5-6),– . , . , . X-gal.
- . , .
 , (1-4) (5-6)
, (1-4) (5-6):
50 96- 20 , sfGFP. ( , ) Transcriptic
Tecan Infinite .
, , , sfGFP. , , - , sfGFP . , sfGFP, , , , .
(OD600) 20 ( 60 ).
for t in [0,4,8,12,16,20]: abs_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_abs_{}.csv".format(t), index_col="Well") flr_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_fl2_{}.csv".format(t), index_col="Well") if t == 0: new_data = abs_data.join(flr_data) else: new_data = new_data.join(abs_data, rsuffix='_{}'.format(t)) new_data = new_data.join(flr_data, rsuffix='_{}'.format(t)) new_data.columns = ["OD 600:nanometer_0", "Fluorescence_0"] + list(new_data.columns[2:])
Nous plaçons sur la carte les données de la 20ème heure et les traces des mesures précédentes. En fait, je ne suis intéressé que par les dernières données, car c'est alors qu'il faut observer un pic de fluorescence. svg = [] W, H = 800, 500 min_x, max_x = 0, 0.8 min_y, max_y = 0, 50000 def _toxy(x, y): return W*(x-min_x)/(max_x-min_x), HH*(y-min_y)/(max_y-min_y) def _topt(x, y): return ','.join(map(str,_toxy(x,y))) ab_fls = [[row[0]] + [list(row[1])] for row in new_data.iterrows()]
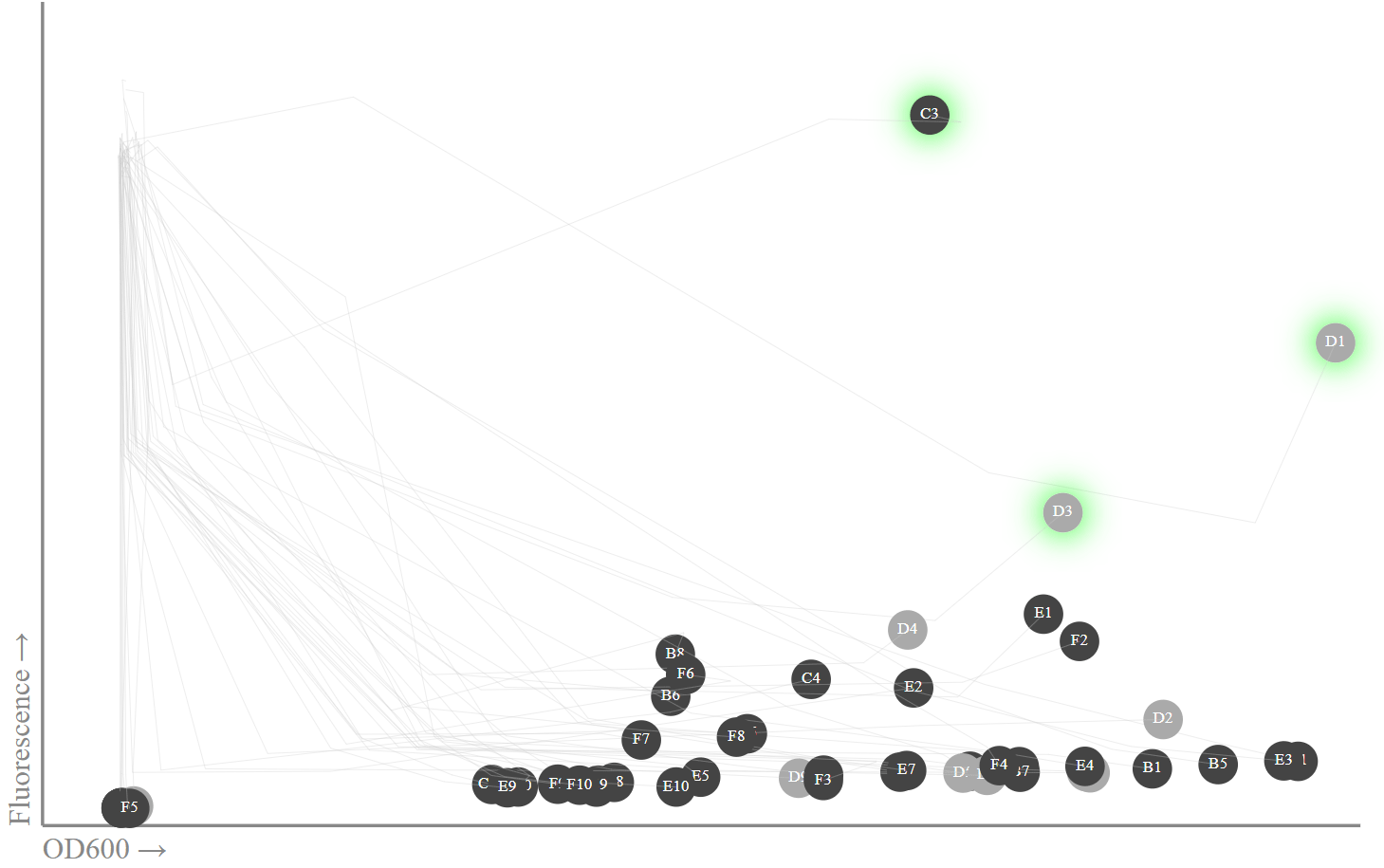 OD600: , . , sfGFPminiprep
OD600: , . , sfGFPminiprep , , 13. , - miniprep - Transcriptic, . (C1, D1, D3) (B1, B3, E1), sfGFP
muscle .
C1, D3 D3 sfGFP, B1, B3 E1 .
, . , 0 (40 000 ). 20- OD600 (, - ), . , , , , 11-15 .
(. . , ), , , ).
, , 50 sfGFP . , . , ( , , miniprep), 200 , .
, . , GFP, Python!
:
Prix
, , $360, :
- $70
- $32 PCR
- $31
- $32
- $53
- $67
- $75 3 miniprep'
, $250-300 . , 50 , , .
, ( ) ( IT). Transcriptic , . , , . , , , , .
, . , - , . , : , , IDT .
:
, . , :
- ! , . autoprotocol, .
- . 100 , .
- , , PCR. , , ? / ? , , , « 2-3 ». ?
- . . , .
- . .
- . . , 1 96 (96−x) 96- , .
- . csv , .
- . - , .
, , , . , 1994 :
- Transcriptic — . , , , . , , .
- — Transcriptic.
- , . Transcriptic ( , , ).
- , ( : ~$0). , .
- Transcriptic . , , .
, , - , , .
, :
- Twist/IDT/Gen9 Transcriptic (, - ).
- , , , , . .
- ( NEB, IDT) (, primer3 ).
(
) , ,
. , in vivo (. . ).
, , : RBS , ; ; .
Pourquoi tout ça?
, . :
- - //, .
- , , .
- in vivo split-GFP .
- scFv . scFvs - .
- BiTE , ( , ).
- , ( ).
- , . 1000 10 000 ? , GFP?
, ,
iGEM .
Transcriptic .