Comment lire cet article : Je m'excuse du fait que le texte s'est avéré si long et chaotique. Pour vous faire gagner du temps, je commence chaque chapitre par l'introduction de «Ce que j'ai appris», dans lequel j'explique l'essence du chapitre en une ou deux phrases.
"Montrez simplement la solution!" Si vous voulez simplement voir où je suis arrivé, alors allez au chapitre "Devenez plus inventif", mais je pense que c'est plus intéressant et utile de lire sur les échecs.Récemment, j'ai été chargé de mettre en place un processus de traitement d'un grand volume des séquences d'ADN d'origine (techniquement, il s'agit d'une puce SNP). Il était nécessaire d'obtenir rapidement des données sur un emplacement génétique donné (appelé SNP) pour la modélisation ultérieure et d'autres tâches. Avec l'aide de R et AWK, j'ai pu nettoyer et organiser les données de manière naturelle, accélérant considérablement le traitement des demandes. Cela n'a pas été facile pour moi et a nécessité de nombreuses itérations. Cet article vous aidera à éviter certaines de mes erreurs et à montrer ce que j'ai fait au final.
Tout d'abord, quelques explications introductives.
Les données
Notre centre de traitement des informations génétiques de l'université nous a fourni 25 To de données TSV. Je les ai divisés en 5 paquets compressés par Gzip, chacun contenant environ 240 fichiers de quatre gigaoctets. Chaque ligne contenait des données pour un SNP d'une personne. Au total, des données sur environ 2,5 millions de SNP et environ 60 000 personnes ont été transmises. En plus des informations SNP, il y avait de nombreuses colonnes dans les fichiers avec des nombres reflétant diverses caractéristiques, telles que l'intensité de lecture, la fréquence des différents allèles, etc. Il y avait environ 30 colonnes avec des valeurs uniques.
But
Comme pour tout projet de gestion des données, le plus important était de déterminer comment les données seraient utilisées. Dans ce cas,
pour la plupart, nous sélectionnerons des modèles et des workflows pour SNP basés sur SNP . Autrement dit, nous aurons besoin en même temps de données pour un seul SNP. J'ai dû apprendre à extraire tous les enregistrements liés à l'un des 2,5 millions de SNP aussi simplement que possible, plus rapidement et moins cher.
Comment ne pas le faire
Je citerai un cliché approprié:
Je n'ai pas échoué mille fois, je viens de découvrir mille façons de ne pas analyser un tas de données dans un format pratique pour les requêtes.
Première tentative
Ce que j'ai appris : il n'y a pas de moyen bon marché d'analyser 25 To à la fois.
Après avoir écouté le sujet «Advanced Big Data Processing Methods» à l'Université Vanderbilt, j'étais sûr que c'était un chapeau. Il faudra peut-être une heure ou deux pour configurer le serveur Hive pour qu'il exécute toutes les données et rende compte du résultat. Étant donné que nos données sont stockées dans AWS S3, j'ai utilisé le service
Athena , qui vous permet d'appliquer des requêtes Hive SQL aux données S3. Pas besoin de configurer / augmenter le cluster Hive, et même de payer uniquement pour les données que vous recherchez.
Après avoir montré à Athena mes données et leur format, j'ai effectué quelques tests avec des requêtes similaires:
select * from intensityData limit 10;
Et a rapidement obtenu des résultats bien structurés. C'est fait.
Jusqu'à ce que nous essayions d'utiliser les données dans le travail ...
On m'a demandé de retirer toutes les informations SNP afin de tester le modèle dessus. J'ai lancé une requête:
select * from intensityData where snp = 'rs123456';
... et attendu. Après huit minutes et plus de 4 To des données demandées, j'ai obtenu le résultat. Athena facture des frais pour la quantité de données trouvées, à 5 $ par téraoctet. Cette seule demande a donc coûté 20 $ et huit minutes d'attente. Pour exécuter le modèle selon toutes les données, il a fallu attendre 38 ans et payer 50 millions de dollars, ce qui ne nous convenait évidemment pas.
Il fallait utiliser Parquet ...
Ce que j'ai appris : soyez prudent avec la taille de vos dossiers de parquet et leur organisation.
Au début, j'ai essayé de corriger la situation en convertissant tous les TSV en
fichiers Parquet . Ils sont pratiques pour travailler avec de grands ensembles de données, car les informations qu'ils contiennent sont stockées sous forme de colonnes: chaque colonne se trouve dans son propre segment mémoire / disque, contrairement aux fichiers texte dans lesquels les lignes contiennent des éléments de chaque colonne. Et si vous avez besoin de trouver quelque chose, alors lisez simplement la colonne nécessaire. De plus, une plage de valeurs est stockée dans chaque fichier d'une colonne, donc si la valeur souhaitée ne se trouve pas dans la plage de colonnes, Spark ne perdra pas de temps à analyser le fichier entier.
J'ai exécuté une tâche
AWS Glue simple pour convertir nos TSV en Parquet et j'ai déposé de nouveaux fichiers dans Athena. Cela a pris environ 5 heures. Mais lorsque j'ai lancé la demande, il a fallu environ le même temps et un peu moins d'argent pour la compléter. Le fait est que Spark, essayant d'optimiser la tâche, a simplement déballé un bloc TSV et l'a placé dans son propre bloc Parquet. Et comme chaque bloc était suffisamment grand et contenait les enregistrements complets de nombreuses personnes, tous les SNP étaient stockés dans chaque fichier, donc Spark a dû ouvrir tous les fichiers pour extraire les informations nécessaires.
Curieusement, le type de compression par défaut (et recommandé) dans Parquet - snappy - n'est pas séparable. Par conséquent, chaque exécuteur s'est attaché à la tâche de décompresser et de télécharger l'ensemble de données complet de 3,5 Go.

Nous comprenons le problème
Ce que j'ai appris : le tri est difficile, surtout si les données sont distribuées.
Il me semblait que maintenant je comprenais l'essence du problème. Tout ce que j'avais à faire était de trier les données par colonne SNP, pas par personnes. Ensuite, plusieurs SNP seront stockés dans un bloc de données séparé, puis la fonction intelligente Parquet "ouverte uniquement si la valeur est dans la plage" apparaîtra dans toute sa splendeur. Malheureusement, trier des milliards de lignes dispersées sur un cluster s'est avéré être une tâche ardue.
AWS ne veut certainement pas rendre l'argent à cause de "Je suis un étudiant distrait." Après avoir commencé à trier sur Amazon Glue, cela a fonctionné pendant 2 jours et s'est écrasé.
Et le partitionnement?
Ce que j'ai appris : les partitions dans Spark doivent être équilibrées.
Puis l'idée m'est venue de partitionner les données sur les chromosomes. Il y en a 23 (et quelques autres, étant donné l'ADN mitochondrial et les zones non cartographiées).
Cela vous permettra de diviser les données en portions plus petites. Si vous ajoutez une seule ligne
partition_by = "chr" à la fonction d'exportation Spark dans le script Glue, les données doivent être triées dans des compartiments.
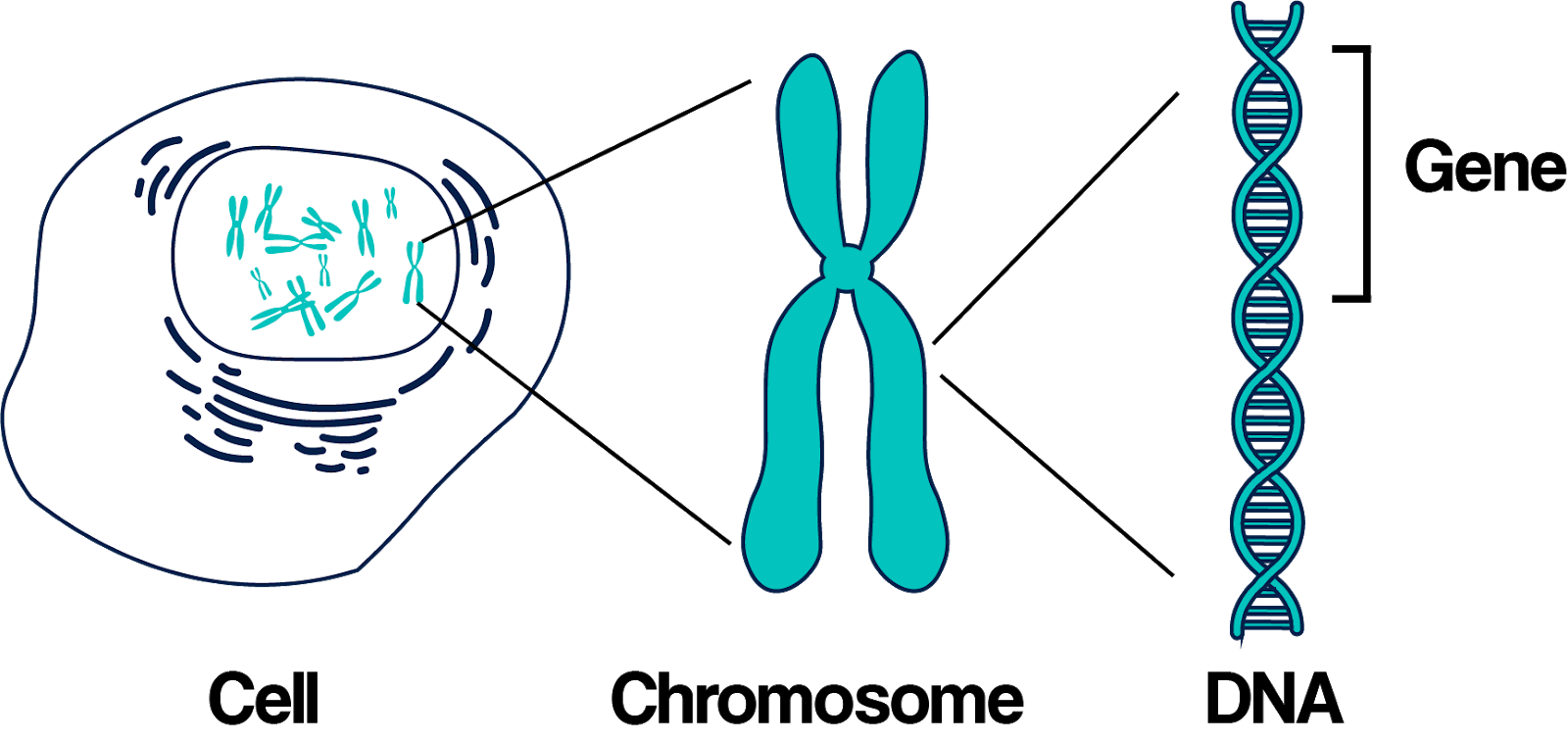 Le génome se compose de nombreux fragments appelés chromosomes.
Le génome se compose de nombreux fragments appelés chromosomes.Malheureusement, cela n'a pas fonctionné. Les chromosomes ont des tailles différentes, et donc une quantité d'informations différente. Cela signifie que les tâches envoyées par Spark aux travailleurs n'étaient pas équilibrées et exécutées lentement, car certains nœuds se terminaient plus tôt et étaient inactifs. Cependant, les tâches ont été achevées. Mais lors de la demande d'un SNP, le déséquilibre a de nouveau causé des problèmes. Le coût du traitement des SNP sur des chromosomes plus gros (c'est-à-dire d'où nous voulons obtenir les données) n'a diminué que d'environ 10 fois. Beaucoup, mais pas assez.
Et si vous vous divisez en partitions encore plus petites?
Ce que j'ai appris : n'essayez jamais de faire 2,5 millions de partitions.
J'ai décidé de me promener et de partitionner chaque SNP. Cela garantissait la même taille de partitions.
MAUVAIS ÉTAIT UNE IDÉE . J'ai profité de Glue et ajouté la ligne innocente
partition_by = 'snp' . La tâche a commencé et a commencé à s'exécuter. Un jour plus tard, j'ai vérifié et vu que rien n'était écrit en S3 jusqu'à présent, alors j'ai tué la tâche. Il semble que Glue écrivait des fichiers intermédiaires dans un endroit caché dans S3, et beaucoup de fichiers, peut-être quelques millions. En conséquence, mon erreur a coûté plus de mille dollars et n'a pas plu à mon mentor.
Partitionnement + tri
Ce que j'ai appris : le tri est toujours difficile, tout comme la configuration de Spark.
La dernière tentative de partitionnement a consisté à partitionner les chromosomes puis à trier chaque partition. En théorie, cela accélérerait chaque demande, car les données SNP souhaitées devraient se trouver dans plusieurs morceaux de parquet dans une plage donnée. Hélas, le tri des données même partitionnées s'est avéré être une tâche difficile. En conséquence, je suis passé à EMR pour un cluster personnalisé et j'ai utilisé huit instances puissantes (C5.4xl) et Sparklyr pour créer un flux de travail plus flexible ...
# Sparklyr snippet to partition by chr and sort w/in partition # Join the raw data with the snp bins raw_data group_by(chr) %>% arrange(Position) %>% Spark_write_Parquet( path = DUMP_LOC, mode = 'overwrite', partition_by = c('chr') )
... cependant, la tâche n'était toujours pas terminée. J'ai réglé chaque chose: j'ai augmenté l'allocation de mémoire pour chaque exécuteur de requête, utilisé des nœuds avec une grande quantité de mémoire, utilisé des variables de diffusion, mais à chaque fois cela s'est avéré être des demi-mesures, et progressivement les artistes ont commencé à échouer, jusqu'à ce que tout s'arrête.
Je deviens plus inventif
Ce que j'ai appris : parfois, les données spéciales nécessitent des solutions spéciales.
Chaque SNP a une valeur de position. Il s'agit du nombre correspondant au nombre de bases situées le long de son chromosome. Il s'agit d'un moyen bon et naturel d'organiser nos données. Au début, je voulais partitionner par région de chaque chromosome. Par exemple, positions 1 - 2000, 2001 - 4000, etc. Mais le problème est que les SNP ne sont pas distribués également entre les chromosomes, c'est pourquoi la taille des groupes variera considérablement.
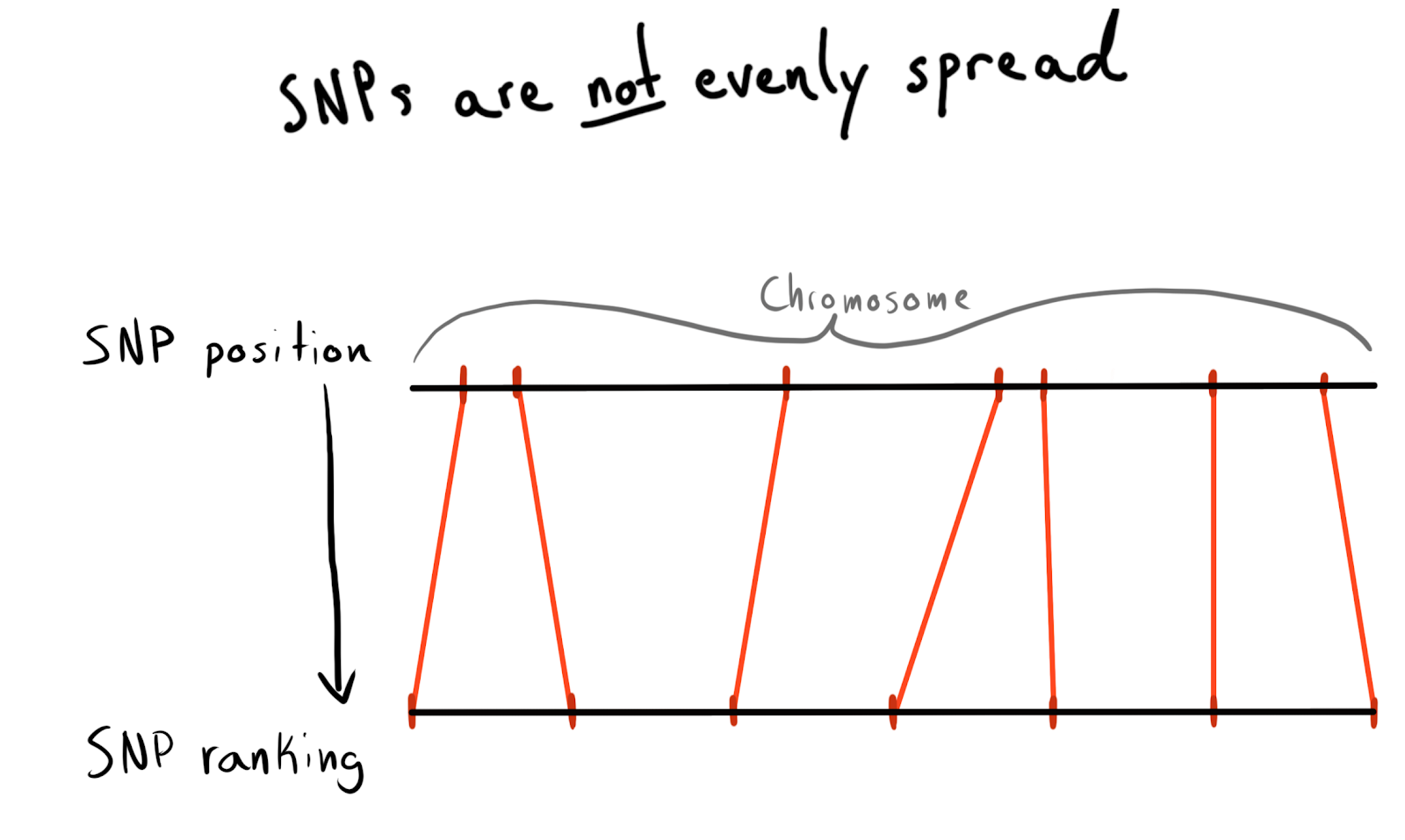
En conséquence, je suis venu à être divisé en catégories (grade) postes. Selon les données déjà téléchargées, j'ai lancé une demande de liste de SNP uniques, de leurs positions et de leurs chromosomes. Il a ensuite trié les données à l'intérieur de chaque chromosome et collecté le SNP en groupes (bac) d'une taille donnée. Dites 1000 SNP chacun. Cela m'a donné une relation SNP avec un groupe en chromosome.
Au final, j'ai fait des groupes (bin) sur 75 SNP, j'expliquerai la raison ci-dessous.
snp_to_bin <- unique_snps %>% group_by(chr) %>% arrange(position) %>% mutate( rank = 1:n() bin = floor(rank/snps_per_bin) ) %>% ungroup()
Essayez d'abord avec Spark
Ce que j'ai appris : l'intégration de Spark est rapide, mais le partitionnement reste cher.
Je voulais lire ce petit bloc de données (2,5 millions de lignes) dans Spark, le combiner avec des données brutes, puis partitionner par la colonne
bin nouvellement ajoutée.
J'ai utilisé
sdf_broadcast() , donc Spark découvre qu'il devrait envoyer une trame de données à tous les nœuds. Ceci est utile si les données sont petites et requises pour toutes les tâches. Sinon, Spark essaie d'être intelligent et distribue les données selon les besoins, ce qui peut provoquer des freins.
Et encore une fois, mon idée n'a pas fonctionné: les tâches ont fonctionné pendant un certain temps, ont terminé la fusion, puis, comme les exécuteurs exécutés par partitionnement, elles ont commencé à échouer.
Ajouter AWK
Ce que j'ai appris : ne dormez pas lorsque les bases vous apprennent. Certes, quelqu'un a déjà résolu votre problème dans les années 1980.
Jusqu'à présent, la cause de tous mes échecs avec Spark était la confusion des données dans le cluster. Peut-être que la situation peut être améliorée par le prétraitement. J'ai décidé d'essayer de diviser les données brutes du texte en colonnes chromosomiques, alors j'espérais fournir à Spark des données «pré-partitionnées».
J'ai cherché sur StackOverflow pour savoir comment décomposer les valeurs des colonnes et j'ai trouvé
une excellente réponse. À l'aide d'AWK, vous pouvez diviser un fichier texte en valeurs de colonne en écrivant dans le script, plutôt qu'en envoyant les résultats à
stdout .
Pour les tests, j'ai écrit un script Bash. J'ai téléchargé l'un des TSV emballés, puis l'ai déballé avec
gzip et l'ai envoyé à
awk .
gzip -dc path/to/chunk/file.gz | awk -F '\t' \ '{print $1",..."$30">"chunked/"$chr"_chr"$15".csv"}'
Ça a marché!
Remplissage de noyau
Ce que j'ai appris : le
gnu parallel est une chose magique, tout le monde devrait l'utiliser.
La séparation a été plutôt lente, et quand j'ai commencé
htop pour tester l'utilisation d'une instance EC2 puissante (et coûteuse), il s'est avéré que j'utilisais un seul cœur et environ 200 Mo de mémoire. Afin de résoudre le problème et de ne pas perdre beaucoup d'argent, il a fallu trouver comment paralléliser le travail. Heureusement, dans l'étonnant
Data Science de Jeron Janssens
sur le livre de la
ligne de commande , j'ai trouvé un chapitre sur la parallélisation. De là, j'ai appris sur
gnu parallel , une méthode très flexible pour implémenter le multithreading sur Unix.
Quand j'ai commencé la partition en utilisant un nouveau processus, tout allait bien, mais il y avait un goulot d'étranglement - le téléchargement d'objets S3 sur le disque n'était pas trop rapide et pas complètement parallélisé. Pour résoudre ce problème, j'ai fait ceci:
- J'ai découvert qu'il est possible d'implémenter l'étape de téléchargement S3 directement dans le pipeline, éliminant complètement le stockage intermédiaire sur disque. Cela signifie que je peux éviter d'écrire des données brutes sur le disque et utiliser un stockage encore plus petit et donc moins cher sur AWS.
- La commande
aws configure set default.s3.max_concurrent_requests 50 considérablement augmenté le nombre de threads que l'AWS CLI utilise (il y en a 10 par défaut).
- Je suis passé à l'instance EC2 optimisée pour la vitesse du réseau, avec la lettre n dans le nom. J'ai constaté que la perte de puissance de calcul lors de l'utilisation de n instances est plus que compensée par une augmentation de la vitesse de téléchargement. Pour la plupart des tâches, j'ai utilisé c5n.4xl.
- J'ai changé
gzip en pigz , c'est un outil gzip qui peut faire des choses sympas pour paralléliser la tâche initialement inégalée de décompresser des fichiers (cela a le moins aidé).
Ces étapes sont combinées entre elles pour que tout fonctionne très rapidement. Grâce à la vitesse de téléchargement accrue et au refus d'écrire sur le disque, je pouvais désormais traiter un paquet de 5 téraoctets en quelques heures seulement.
Ce tweet était censé mentionner «TSV». Hélas.
Utilisation de données analysées à nouveau
Ce que j'ai appris : Spark aime les données non compressées et n'aime pas combiner les partitions.
Maintenant, les données étaient en S3 dans un format décompressé (lu, partagé) et semi-ordonné, et je pouvais revenir à Spark à nouveau. Une surprise m'attendait: je n'ai de nouveau pas réussi à atteindre le résultat souhaité! Il était très difficile de dire à Spark exactement comment les données étaient partitionnées. Et même quand je l'ai fait, il s'est avéré qu'il y avait trop de partitions (95 000), et quand j'ai réduit leur nombre à des limites cohérentes avec
coalesce , cela a ruiné mon partitionnement. Je suis sûr que cela peut être corrigé, mais en quelques jours de recherche, je n'ai pas pu trouver de solution. Au final, j'ai terminé toutes les tâches dans Spark, même si cela a pris du temps, et mes fichiers de parquet fractionnés n'étaient pas très petits (~ 200 Ko). Cependant, les données étaient là où elles étaient nécessaires.
 Trop petit et différent, merveilleux!
Trop petit et différent, merveilleux!Test des demandes Spark locales
Ce que j'ai appris : Spark a trop de temps pour résoudre des problèmes simples.
En téléchargeant les données dans un format intelligent, j'ai pu tester la vitesse. J'ai configuré un script sur R pour démarrer le serveur Spark local, puis j'ai chargé la trame de données Spark à partir du référentiel spécifié des groupes de parquet (bin). J'ai essayé de charger toutes les données, mais je n'ai pas pu faire en sorte que Sparklyr reconnaisse le partitionnement.
sc <- Spark_connect(master = "local") desired_snp <- 'rs34771739' # Start a timer start_time <- Sys.time() # Load the desired bin into Spark intensity_data <- sc %>% Spark_read_Parquet( name = 'intensity_data', path = get_snp_location(desired_snp), memory = FALSE ) # Subset bin to snp and then collect to local test_subset <- intensity_data %>% filter(SNP_Name == desired_snp) %>% collect() print(Sys.time() - start_time)
L'exécution a duré 29,415 secondes. Beaucoup mieux, mais pas trop bon pour tester quoi que ce soit en masse. De plus, je n'ai pas pu accélérer le travail avec la mise en cache, car lorsque j'essayais de mettre en cache la trame de données en mémoire, Spark se bloquait toujours, même lorsque j'allouais plus de 50 Go de mémoire pour un ensemble de données pesant moins de 15.
Retour à AWK
Ce que j'ai appris : les tableaux associatifs AWK sont très efficaces.
J'ai compris que je pouvais atteindre une vitesse plus élevée. Je me suis rappelé que dans l’excellent guide AWK de
Bruce Barnett , j’ai lu sur une fonctionnalité intéressante appelée «
tableaux associatifs ». En fait, ce sont des paires clé-valeur, qui pour une raison quelconque étaient appelées différemment dans AWK, et donc je ne les ai pas particulièrement mentionnées.
Roman Cheplyaka a rappelé que le terme «tableaux associatifs» est beaucoup plus ancien que le terme «paire clé-valeur». Même si vous
recherchez une valeur-clé dans Google Ngram , vous ne verrez pas ce terme là, mais vous trouverez des tableaux associatifs! De plus, la paire clé-valeur est le plus souvent associée aux bases de données, il est donc beaucoup plus logique de comparer avec hashmap. J'ai réalisé que je pouvais utiliser ces tableaux associatifs pour connecter mes SNP à la table bin et aux données brutes sans utiliser Spark.
Pour cela, dans le script AWK, j'ai utilisé le bloc
BEGIN . Il s'agit d'un morceau de code qui est exécuté avant le transfert de la première ligne de données vers le corps principal du script.
join_data.awk BEGIN { FS=","; batch_num=substr(chunk,7,1); chunk_id=substr(chunk,15,2); while(getline < "snp_to_bin.csv") {bin[$1] = $2} } { print $0 > "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv" }
La commande
while(getline...) chargé toutes les lignes du groupe CSV (bin), définissez la première colonne (nom SNP) comme clé pour le tableau associatif
bin et la deuxième valeur (group) comme valeur. Ensuite, dans le bloc
{ } , qui est appliqué à toutes les lignes du fichier principal, chaque ligne est envoyée au fichier de sortie, qui obtient un nom unique en fonction de son groupe (bin):
..._bin_"bin[$1]"_...Les
chunk_id batch_num et
chunk_id correspondaient aux données fournies par le pipeline, ce qui évitait l'état de course, et chaque thread d'exécution lancé en
parallel écrivait dans son propre fichier unique.
Depuis que j'ai dispersé toutes les données brutes dans des dossiers sur les chromosomes laissés après ma précédente expérience avec AWK, je pouvais maintenant écrire un autre script Bash à traiter sur le chromosome à la fois et donner des données partitionnées plus profondes à S3.
DESIRED_CHR='13'
Le script a deux sections
parallel .
Dans la première section, les données de tous les fichiers contenant des informations sur le chromosome souhaité sont lues, puis ces données sont distribuées en flux qui dispersent les fichiers dans les groupes correspondants (bin). Pour éviter que des conditions de
chr_10_bin_52_batch_2_aa.csv ne se produisent lorsque plusieurs flux sont écrits dans le même fichier, AWK transfère les noms de fichier pour écrire des données à différents endroits, par exemple,
chr_10_bin_52_batch_2_aa.csv . En conséquence, de nombreux petits fichiers sont créés sur le disque (pour cela, j'ai utilisé des volumes EBS de téraoctets).
Le pipeline de la deuxième section
parallel passe par les groupes (bin) et combine leurs fichiers individuels en CSV communs avec
cat , puis les envoie pour exportation.
Diffuser vers R?
Ce que j'ai appris : vous pouvez accéder à
stdin et
stdout partir d'un script R, et donc l'utiliser dans le pipeline.
Dans le script Bash, vous remarquerez peut-être cette ligne:
...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... Il traduit tous les fichiers de groupe concaténés (bin) dans le script R ci-dessous.
{} est une technique
parallel spéciale qui insère toutes les données envoyées par elle dans le flux spécifié directement dans la commande elle-même. L'option
{#} fournit un ID de thread unique et
{%} représente le numéro de l'emplacement de travail (répété, mais jamais en même temps). Une liste de toutes les options se trouve dans la
documentation. #!/usr/bin/env Rscript library(readr) library(aws.s3) # Read first command line argument data_destination <- commandArgs(trailingOnly = TRUE)[1] data_cols <- list(SNP_Name = 'c', ...) s3saveRDS( read_csv( file("stdin"), col_names = names(data_cols), col_types = data_cols ), object = data_destination )
Lorsque la variable de
file("stdin") est transmise à
readr::read_csv , les données traduites dans le script R sont chargées dans le cadre, qui est ensuite écrit directement dans S3 en tant que fichier
.rds à l'aide de
aws.s3 .
RDS est un peu comme une version plus récente de Parquet, sans les fioritures du stockage sur colonne.
Après avoir terminé le script Bash, j'ai reçu un
.rds fichiers
.rds trouvant dans S3, ce qui m'a permis d'utiliser une compression efficace et des types intégrés.
Malgré l'utilisation du frein R, tout a fonctionné très rapidement. Il n'est pas surprenant que les fragments sur R qui sont responsables de la lecture et de l'écriture des données soient bien optimisés. Après avoir testé sur un chromosome de taille moyenne, la tâche s'est terminée sur l'instance C5n.4xl en environ deux heures.
Limitations de S3
Ce que j'ai appris : grâce à l'implémentation intelligente des chemins, S3 peut traiter de nombreux fichiers.
J'étais inquiet si S3 pouvait gérer un grand nombre de fichiers qui y étaient transférés. Je pourrais donner un sens aux noms de fichiers, mais comment S3 les recherchera-t-il?
 S3 ,
S3 , / . FAQ- S3., S3 - . (bucket) , — .
Amazon, , «-----» . : get-, . , 20 . bin-. , , (, , ). .
?
: — .
: « ?» ( gzip CSV- 7 ) . , R Parquet ( Arrow) Spark. R, , , .
: , .
, .
EC2 , ( , Spark ). , , AWS- 10 .
R .
S3 , .
library(aws.s3) library(tidyverse) chr_sizes <- get_bucket_df( bucket = '...', prefix = '...', max = Inf ) %>% mutate(Size = as.numeric(Size)) %>% filter(Size != 0) %>% mutate(
, , ,
num_jobs , .
num_jobs <- 7
purrr .
1:1000 %>% map_df(shuffle_job) %>% filter(sd == min(sd)) %>% pull(data) %>% pluck(1)
, . Bash-
for . 10 . , . , .
for DESIRED_CHR in "16" "9" "7" "21" "MT" do
:
sudo shutdown -h now
… ! AWS CLI
user_data Bash- . , .
aws ec2 run-instances ...\ --tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]" \ --user-data file://<<job_script_loc>>
!
: API .
- . , . API .
.rds Parquet-, , . R-.
, ,
get_snp .
pkgdown , .
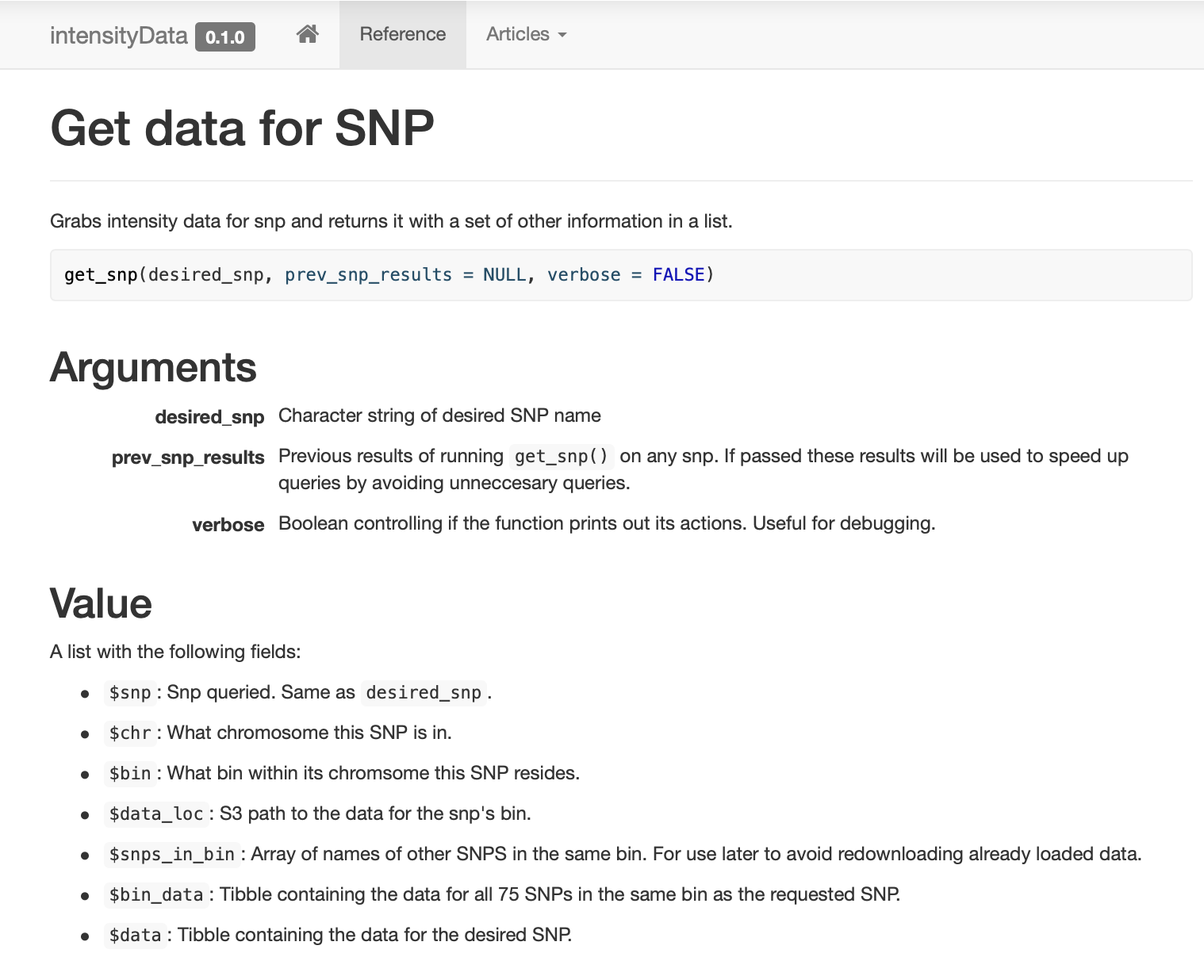
: , !
SNP , (binning) . SNP, (bin). ( ) .
, . , . ,
dplyr::filter , , .
,
prev_snp_results snps_in_bin . SNP (bin), , . SNP (bin) :
Résultats
( ) , . , . .
, , , …
. . ( ), , (bin) , SNP 0,1 , , S3 .
Conclusion
— . , . , . , , , . , , , , . , , , , - .
. , , «» , . .
:
- 25 ;
- Parquet- ;
- Spark ;
- 2,5 ;
- , Spark;
- ;
- Spark , ;
- , , - 1980-;
gnu parallel — , ;
- Spark ;
- Spark ;
- AWK ;
stdin stdout R-, ;
- S3 ;
- — ;
- , ;
- API ;
- , !