Dans l'
article détaillé précédent sur le
génome complet, nous avions promis de publier trois problèmes et de donner un test à celui qui résoudrait tous les trois correctement. Dans le même temps, nous donnons des exemples de la façon de travailler avec des données génétiques dans ces tâches. Aujourd'hui, nous publions le premier.

Dans le premier
article, nous avons partagé des informations et des liens utiles qui sont utiles pour travailler avec des données bioinformatiques. Nous vous recommandons de le lire en premier si vous l'avez manqué.
Clause de non-responsabilitéLe travail avec les données génétiques est effectué sur les systèmes Unix (Linux, macOS), car certaines commandes et logiciels ne sont pas disponibles sur Windows. Par conséquent, pour les utilisateurs de Windows, l'une des solutions les plus simples consiste à louer une machine virtuelle Linux.
Toutes les opérations décrites ci-dessous sont effectuées sur la ligne de commande - terminal. Avant de commencer, découvrez comment travailler dans un terminal exécutant votre système d'exploitation et utilisez des commandes, car certaines d'entre elles peuvent potentiellement endommager le système d'exploitation et vos données.
Logiciels requis
Nous avons collecté l'
image d'une machine virtuelle (VM) avec tous les logiciels nécessaires sur Yandex.Cloud. Inscrivez-vous dans Yandex.Cloud, dans votre compte dans la section Compute Cloud, cliquez sur Créer une machine virtuelle. En tant qu'image publique, sélectionnez 1000 génomes dans le catalogue Atlas Data Analysis.
Configuration VM: 100% 2vCPU, 8 Go de RAM, 20 Go de disque dur. Lors de la création d'une machine virtuelle, vous devez saisir les données de paiement, mais rien n'est radié du compte. Un démarrage et une subvention supplémentaire sur un mot de code suffisent pour travailler gratuitement avec une machine virtuelle et une image d'Atlas jusqu'au 31 décembre 2019. Pour recevoir une subvention pour l'exécution de tâches, envoyez le mot de code "ATLAS" au
support Yandex.Cloud .
Remarque: la subvention est valable pour les nouveaux utilisateurs de Yandex.Cloud qui se sont inscrits depuis le 18 décembre 2019 ou pour ceux qui ont encore une période d'essai et ont une subvention de départ. Le mot de code ATLAS n'est valable qu'une seule fois.
Créez d'abord une clé ssh sur l'ordinateur local à partir duquel vous prévoyez de vous connecter à la machine virtuelle:
ssh-keygen -o -t rsa -b 4096 -C "my-local-machine" -f ~/.ssh/yandex-cloud -a 100
N'oubliez pas de copier le contenu du fichier
~/.ssh/yandex-cloud.pub dans la fenêtre appropriée lors de la création de la machine virtuelle.
Si vous souhaitez installer le logiciel sur votre ordinateur, voici toutes les informations d'installation. Si vous décidez d'utiliser Yandex.Cloud, créez une machine virtuelle et passez à la section suivante.
Plink
Plink est un progiciel de manipulation de données génétiques et de recherche d'association à large génome (GWAS). Il a été développé par le généticien Sean Purcell (Shaun Purcell). Depuis 2008, avec l'aide de Plink, des centaines de GWAS ont été réalisés dans le monde, dont les meilleurs résultats qu'Atlas utilise comme source de données pour les algorithmes de calcul des risques de maladie.
Plink propose un ensemble d'outils pour stocker et convertir les données de génotypage et les rechercher. Plink permet également le traitement statistique, l'analyse de déséquilibre de liaison (LD), l'analyse d'identité par descendance (IBD) et d'identité (état par IBS), la stratification de la population et les tests d'épistase - l'interaction de plusieurs variations génétiques entre eux.
L'IBD et l'IBS sont utilisés pour analyser la composition de la population et déterminer la parenté.
Un exemple d'épistase est la variation de rs7412 et rs429358 dans le gène APOE, une certaine combinaison de variantes dont augmente fortement le risque de développer la maladie d'Alzheimer, tandis que chaque variante individuellement ne contribue que faiblement au risque.
Téléchargez la version stable de Plink sur le site
officiel .
BCFtools
BCFtools est un ensemble d'utilitaires pour manipuler des données génétiques au format VCF et son équivalent binaire BCF. La liste des applications possibles de BCFtools comprend l'annotation, le filtrage, la fusion et le fractionnement des fichiers VCF / BCF, la recherche de leurs intersections, l'indexation, la recherche sélective, le tri, le comptage des statistiques, etc.
Pour installer, faites:
git clone git://github.com/samtools/htslib.git git clone git://github.com/samtools/bcftools.git cd bcftools
Le processus d'installation est décrit plus en détail
ici .
ROI
Le package KING (Kinship-based INference for Gwas) est utilisé dans les études de population lorsque vous travaillez avec les données d'une recherche d'association à l'échelle du génome pour déterminer les relations familiales dans les données étudiées. Dans cette tâche, KING aidera à déterminer le degré de parenté de plusieurs échantillons du projet 1000 Genomes.
Vous pouvez le télécharger
ici . Pour résoudre les problèmes, le manuel KING est disponible
ici .
Presque toutes les erreurs pouvant survenir pendant le travail avec les outils sont décrites sur Stackoverflow ou son équivalent bioinformatique - Biostars .
Données utilisées
À titre indicatif, nous utilisons les données ouvertes
du projet 1000 Genomes. Pour l'analyse, nous avons sélectionné 10 échantillons avec des informations sur les génotypes d'environ 85 millions de variations obtenues en analysant les données NGS alignées avec la version du génome de référence GRCh37. Les relations familiales et les échantillons de population sont illustrés à la figure 1.
 Figure 1
Figure 1 Pedigree utilisé dans les échantillons VCF. Le carré correspond au sexe masculin, le cercle à la femelle. La ligne pointillée signifie une parenté indéterminée de second ordre.
Prenez note
Le format VCF vous permet de stocker des informations sur le champ d'une personne sous la forme d'un numéro unique, si ces informations étaient connues lors de la génération de VCF. Il ressemble à ceci: le champ GT (génotype, génotype) pour les enregistrements du chromosome X contient une valeur numérique qui correspond à un allèle, pour les hommes et deux pour les femmes. S'il n'y a aucune information sur le champ biologique de l'échantillon séquencé, le champ GT contiendra par défaut deux valeurs numériques (surlignées en rouge sur la figure 2).
Dans les fichiers VCF utilisés dans ce manuel, le chromosome Y est exclu, mais la présence du chromosome Y dans le fichier VCF ne signifie pas toujours que l'échantillon séquencé l'a vraiment. Cela est dû aux régions pseudo-autosomiques (PAR), identiques pour les chromosomes X et Y et situées à leurs extrémités.
Différents chromosomes n'ont normalement pas de régions identiques (homologues) longues, cependant, les chromosomes X et Y ont de telles régions plusieurs millions de paires de bases de long au tout début (PAR1) et à la fin (PAR2). Par conséquent, lors de l'analyse des données NGS chez les hommes dans les régions PAR, deux allèles sont trouvés (un pour chaque chromosome sexuel), et chez les femmes, des génotypes peuvent apparaître dans les régions PAR du chromosome Y, bien qu'en fait ce soient des génotypes de leur chromosome X.
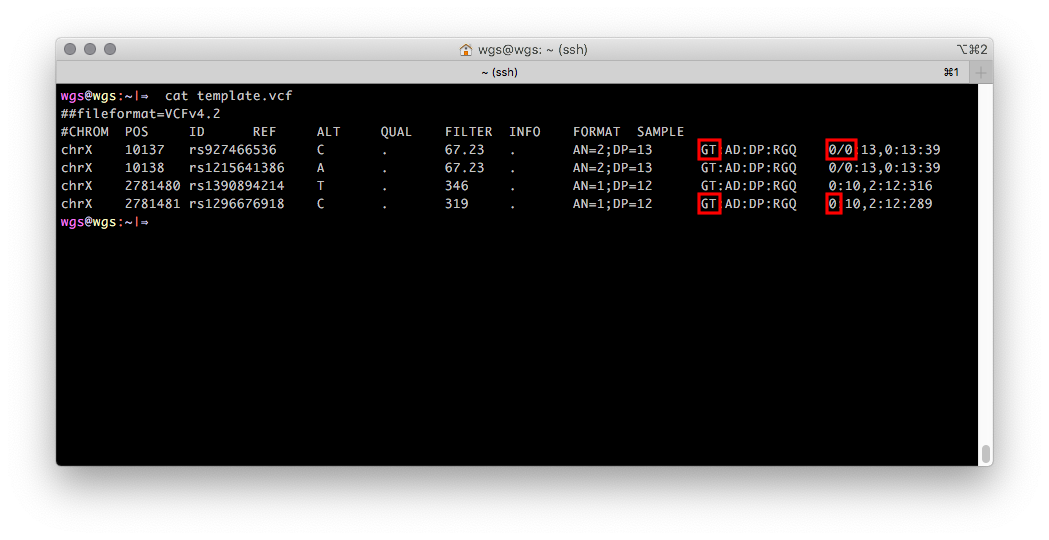 Figure 2
Figure 2 Fichier VCF avec des génotypes du chromosome X d'un homme de la région PAR1 (deux premières entrées) et de la région non pseudo-autosomique (deux dernières entrées).
Unité pédagogique
Le genre génétique est un ensemble de chromosomes sexuels correspondant à la manifestation de caractéristiques sexuelles primaires et secondaires de type masculin ou féminin. Normalement, les hommes ont un chromosome X et un chromosome Y, tandis que les femmes ont deux chromosomes X. Avec divers troubles de la formation des cellules germinales, des œufs et du sperme, un enfant avec un excellent ensemble de chromosomes sexuels peut naître de ses parents, ce qui conduit souvent à des troubles du développement caractéristiques sexuelles primaires et secondaires.
Les deux anomalies sexuelles chromosomiques les plus courantes sont le syndrome de Turner (un ensemble de chromosomes X0, c'est-à-dire un seul chromosome X) et le syndrome de Klinefelter (un ensemble de chromosomes XXY).
Un allèle est un ou plusieurs nucléotides situés à n'importe quelle position dans le génome et ayant une alternative. Le concept est utilisé pour décrire les génotypes. Distinguer les allèles de référence des alternatives. Tous sont stockés dans le fichier VCF dans les champs REF et ALT, respectivement.
Déterminer le sexe
Pour les utilisateurs de Yandex.CloudToutes les données pour effectuer les tâches manuelles et indépendantes sont stockées sur Yandex.Cloud en utilisant la structure ci-dessous. Le dossier
Tutorial contient le fichier VCF nécessaire pour terminer le manuel, le dossier
Test pour les tâches indépendantes. Le dossier
Technical contient deux fichiers avec une liste d'identifiants des variations génétiques:
rsids_for_subsetting.txt utilisé dans le manuel et des tâches pour une exécution indépendante,
external_interpretation_rsids.txt pourrait être nécessaire à l'avenir lors de l'acquisition du séquençage à l'échelle du génome dans l'Atlas pour télécharger des données de génotypage vers des services tiers. Le dossier
Tools contient, entre autres, deux scripts utilisés dans les tâches 2 et 3.
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
Un dossier sera créé dans le répertoire
/home de la machine virtuelle Yandex.Cloud, dont le nom correspond au nom d'utilisateur spécifié lors de la création de la machine virtuelle. Copiez tout du répertoire
/home/ubuntu vers votre répertoire via les commandes suivantes:
cd ~ cp -r /home/ubuntu/* ./
Pour le resteLorsque vous travaillez sur un PC personnel, vous pouvez télécharger les fichiers nécessaires pour la première tâche à partir du
lien . L'archive téléchargée prend en charge une structure de stockage de fichiers similaire à celle utilisée sur Yandex.Cloud:
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
atlas_wgs_contest.tar.gz archive
atlas_wgs_contest.tar.gz avec la commande
tar -xvzf atlas_wgs_contest.tar.gz Les fichiers VCF pour effectuer des tâches sous forme non archivée occupent environ 19 gigaoctets chacun, par conséquent, pour économiser de l'espace, nous vous recommandons de travailler uniquement avec des archives. Tous les programmes répertoriés ci-dessus peuvent déjà fonctionner avec des données VCF compressées. De plus, vous n'avez rien à faire.
Pour déterminer le sexe du sujet, vous devez regarder les génotypes sur le chromosome X et exclure les régions PAR1 et PAR2 situées à son début et à sa fin. Ce sont les intervalles des positions 60001–2699520 et 154931044–155260560 dans la version GRCh37 du génome. Si le génotype contient une désignation numérique, il s'agit du sexe biologique masculin, sinon de la femelle. Il convient de garder à l'esprit que la désignation du sexe dans le fichier VCF dépend de la disponibilité des informations sur le domaine biologique lors de la génération du VCF, de sorte que cette approche ne peut pas toujours être utilisée.
Utilisez la commande suivante pour chacun des échantillons de l'ensemble de données. Remplacez l'identifiant de l'échantillon après l'argument
-s :
(/Data/Tutotrial/CEI.1kg.2019.demo.vcf.gz):
Lors de l'exécution des commandes, vous verrez une partie du contenu du fichier VCF pour l'identifiant d'échantillon spécifié. Le
-r chrX:2699521-154931043 dans BCFtools restreint la visualisation du contenu du fichier à la région du chromosome X de la position 2699521 à la position 154931043 (région non PAR sur la figure 3). Ces limites excluent les régions pseudo-autosomiques inutiles dans ce cas (PAR1 et PAR2). À l'aide des valeurs numériques du champ GT, déterminez le sexe de chaque échantillon.
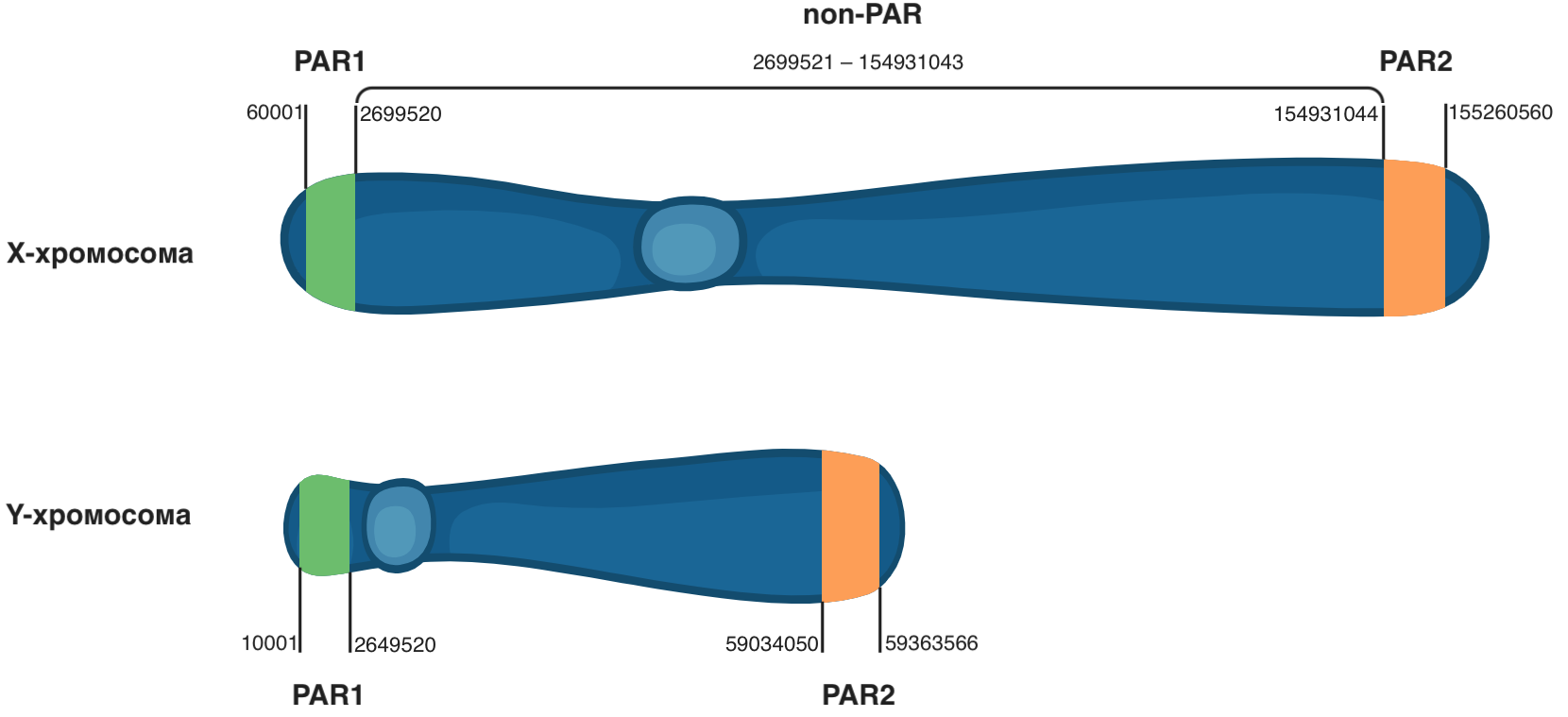 Figure 3
Figure 3 La localisation des régions pseudo-autosomiques de PAR1 et PAR2 sur les chromosomes sexuels.
Vous pouvez voir la liste de tous les exemples d'identificateurs dans le fichier VCF de la figure 1 ou dans la dernière ligne de l'en-tête du fichier VCF. Ils seront répertoriés après le nom de la colonne FORMAT:
Le vrai sexe de ces échantillons est également indiqué dans la figure 1.
Nous déterminons la relation
Pour déterminer la relation, nous devons comparer par paire les données génétiques de tous les échantillons. Il est difficile de le faire en fonction du génome complet: dans ce cas, un fichier VCF prend des dizaines de gigaoctets. Le VCF que nous utilisons ne prend qu'environ 2 gigaoctets, mais nous le filtrons toujours en fonction de la liste des identificateurs de variation génétique (rsID) génotypés sur les puces d'Illumina: GSA v1, GSA v2, HumanOmniExpress v1.0, HumanOmniExpress v1.3, InfiniumExome v1. 1 et Infinium OmniExpressExome v1.4. Ce sont les puces les plus populaires dans le génotypage commercial.
Nous avons compilé une liste de tous les identifiants des variations génétiques de ces puces dans un fichier séparé avec une liste de rsID. Il contient 1,4 million d'identifiants. Pour filtrer le fichier VCF, exécutez la commande suivante:
bcftools view -O z -i 'ID=@rsids_for_subsetting.txt' CEI.1kg.2019.demo.vcf.gz > CEI.1kg.2019.demo.subset.vcf.gz
Chaque fois que vous utilisez BCFtools et d'autres packages pour travailler avec des fichiers VCF, l'historique des commandes précédentes est ajouté à l'en-tête du fichier. Quelle que soit la méthode de filtrage du fichier VCF et les commandes précédemment exécutées, vous pouvez vérifier l'intégrité et l'identité du contenu principal du VCF en calculant la somme de hachage:
La commande
gunzip -c décompresse le fichier et
gunzip -c son contenu dans stdout, à partir de laquelle les lignes d'en-tête du fichier VCF commençant
# sont supprimées (par conséquent, la commande
grep -v "^#" est utilisée). L'en-tête est supprimé afin de comparer l'intégrité des seules données génétiques elles-mêmes, et non les métadonnées sur quels outils et quand ont été utilisés pour travailler avec ce fichier VCF.
Si la valeur de hachage correspond, vous pouvez continuer et convertir VCF au format Plink interne (par défaut, le format Plink est de trois fichiers avec les extensions bed, bim et fam). Dans ces fichiers, il ne reste que le génotype, le chromosome, la position et certaines autres données, et le reste est éliminé. Avec ce format, il est beaucoup plus facile de travailler et de résoudre divers problèmes qui ne nécessitent pas d'informations supplémentaires de VCF. Par exemple, effectuez GWAS.
Cette commande créera trois fichiers dans le dossier:
CEI.1kg.2019.demo.subset.bed
CEI.1kg.2019.demo.subset.bim
CEI.1kg.2019.demo.subset.famVous pouvez déterminer la parenté par paire pour les 10 échantillons. Nous utilisons la commande suivante pour analyser les fichiers Plink:
king -b CEI.1kg.2019.demo.subset.bed --kinship --prefix CEI.1kg.2019.demo.subset.kinship_analysis
Regardez le fichier
CEI.1kg.2019.demo.subset.kinship_analysis.kin0 et faites attention à la colonne Kinship, qui contient les coefficients de parenté pour les paires d'échantillons indiquées respectivement dans ID1 et ID2.
Comparez les coefficients que vous avez obtenus dans le fichier
CEI.1kg.2019.demo.subset.kinship_analysis.kin0 pour toutes les paires d'échantillons avec le pedigree illustré à la figure 1 (la ligne pointillée correspond à la parenté de second ordre, cependant, il n'y a pas de données de parenté exactes, c'est-à-dire il peut y avoir des cousins, tante / neveu ou oncle / nièce). Essayez de tirer votre propre conclusion sur les valeurs des coefficients de parenté qui peuvent correspondre à la parenté de premier et de second ordre.
IndiceExtrait de la documentation KING: les coefficients de parenté> 0,354 correspondent à des échantillons en double ou des jumeaux identiques, de 0,177 à 0,354 à la parenté de premier ordre (parents-enfants, frères et sœurs), de 0,0884 à 0,177 à la parenté de second ordre (cousins, tantes / oncles-neveux), et de 0,0442 à 0,0884 - à la parenté de troisième ordre (grands-parents, petits-enfants, cousins au deuxième degré). Tout ce qui est inférieur à 0,0442 est difficile à interpréter sans ambiguïté.
La première tâche du concours
À l'aide d'un ensemble de données de test de 12 échantillons
Data/Test/CEI.1kg.2019.test.vcf.gz ,
Data/Test/CEI.1kg.2019.test.vcf.gz leur pedigree, guidé par les résultats de la détermination du sexe et de l'analyse de parenté. Les échantillons qui, selon les résultats de l'analyse, ne sont pas en relation avec quelqu'un, sont notés à proximité, sans les relier à une ligne avec d'autres échantillons. Le pedigree peut être composé dans un style similaire à celui de la figure 1, mais cela reste à votre discrétion. Les hommes sont indiqués par un carré, les femmes par un cercle, le mariage par une ligne horizontale, un enfant par une ligne verticale, plusieurs enfants par une ramification horizontale d'une ligne verticale (sous la forme de la lettre P). En savoir plus sur ces désignations
ici .
Comme nous l'avons écrit plus haut, les coefficients de parenté ne peuvent caractériser sans ambiguïté la parenté d'un ordre ou d'un autre: les mêmes coefficients de parenté sont obtenus lors de la comparaison des couples parent-enfant et frère-sœur (parenté de premier ordre). S'il n'est pas possible d'établir la nature de la relation, indiquez l'une des possibilités. Veuillez noter que les échantillons de l'ensemble de données de test ont des identifiants différents de ceux utilisés dans l'ensemble de données d'apprentissage.
Les réponses
doivent être envoyées à
wgs@atlas.ru mail jusqu'au 26 décembre à 23h59. Deux autres tâches seront publiées prochainement et les résultats définitifs de ces tâches seront publiés le 28 décembre. Le gagnant recevra le test du génome complet et les deuxième et troisième places recevront le test génétique Atlas. Il y aura également des prix spéciaux de
Yandex.Cloud . Les anciens et actuels employés d'Atlas ne participent pas au concours;)