Nous continuons le cycle de travail, où nous parlons de la façon de travailler avec les données génétiques. La première
tâche «Découvrir le sexe et le degré de relation» peut déjà être résolue et nous envoyer des réponses. Aujourd'hui, nous publions le second.
Le prix principal est le
génome complet .

Nous avons précédemment partagé des informations et des liens utiles qui peuvent être utiles pour travailler avec des données bioinformatiques. Nous vous recommandons de lire d'abord les articles précédents si vous les avez manqués:
Qu'est-ce que le génome complet et pourquoi est-il nécessaireNuméro de tâche 1. Découvrez le sexe et le degré de relation.Clause de non-responsabilité
Le travail avec les données génétiques est effectué sur les systèmes Unix (Linux, macOS), car certaines commandes et logiciels ne sont pas disponibles sur Windows. Par conséquent, pour les utilisateurs de Windows, l'une des solutions les plus simples consiste à louer une machine virtuelle Linux.
Toutes les opérations décrites ci-dessous sont effectuées sur la ligne de commande - terminal. Avant de commencer, découvrez comment travailler dans un terminal exécutant votre système d'exploitation et utilisez des commandes, car certaines d'entre elles peuvent potentiellement endommager le système d'exploitation et vos données.
Logiciels requis
Nous avons collecté l'
image d'une machine virtuelle (VM) avec tous les logiciels nécessaires sur Yandex.Cloud. Les instructions pour configurer la VM et installer le logiciel se trouvent dans l'
article précédent avec la tâche n ° 1.
Cette fois, vous devrez construire un nuage de points bidimensionnel en utilisant les données obtenues par la méthode d'analyse des principaux composants. Nous vous suggérons de construire ce diagramme en utilisant n'importe quel logiciel qui vous convient: Excel, Google Sheets, Python, R et autres.
Pour terminer la tâche, vous avez besoin du progiciel Plink 1.9. Si vous ne l'avez pas encore installé (et que vous n'avez pas terminé la tâche n ° 1), lisez l'article précédent. Il contient des instructions d'installation. Pour participer au concours du Nouvel An 2019, toutes les tâches doivent être terminées!
Prenez note
L'analyse en composantes principales (ACP) est l'un des algorithmes d'apprentissage automatique sans enseignant lorsqu'une machine recherche indépendamment des modèles dans les données. En génétique, l'ACP permet de regrouper les échantillons en fonction des données de génotypage dans un espace à N dimensions (généralement bidimensionnel), où les principaux composants obtenus expliquent le plus précisément la variabilité des données génétiques d'un échantillon à l'autre.
Lors d'une telle analyse, les échantillons d'une population forment généralement un groupe dont la taille et la fluidité des limites dépendent de la similitude des échantillons au sein d'une population donnée. L'algorithme est susceptible d'identifier des échantillons de différentes populations dans différents clusters. Et des échantillons de populations proches appartenant à la même superpopulation, par exemple, EAS - Asie de l'Est, comme dans la figure 1, seront identifiés les uns près des autres ou même dans des grappes qui se croisent.
 Figure 1
Figure 1 Le pedigree des échantillons utilisés dans le VCF (le carré correspond au sexe masculin, le cercle à la femelle). La ligne pointillée correspond à une relation de second ordre indéterminée.
Une analyse similaire est utilisée pour déterminer la population par génotypage. Pour cela, un ensemble de données de référence est nécessaire, qui se compose d'échantillons dont l'origine est déjà connue. La conclusion sur la population peut être tirée par quel groupe d'échantillons connus est le plus proche des données étudiées.
Pour simplifier, l'essence de l'analyse PCA est que les distances par paires entre les points dans l'espace multidimensionnel sont connues, et ces points doivent être situés dans un espace de plus petite dimension afin que les nouvelles distances par paires soient minimalement différentes des originales. La réduction des dimensions simplifie l'analyse des données, mais plus nous les réduisons, plus les nouvelles distances entre les points diffèrent de l'original. Par conséquent, la tâche de l'analyse PCA implique également de trouver un compromis entre la précision et la facilité d'analyse. Tout est comme dans la vie.
Le mode de réalisation le plus simple de la mise en œuvre de l'ACP sur les données génétiques est basé sur l'identité de certains allèles, qui peuvent être divisés en deux sous-types: IBS (identité par état) et IBD (identité par descendance). IBS signifie l'identité de certains allèles chez deux personnes, mais n'implique pas nécessairement le fait d'une relation entre eux. L'IBD, au contraire, parle de l'identité des allèles en raison de la présence d'un ancêtre commun et, par conséquent, de la parenté.
Les allèles IBD sont sans équivoque des allèles IBS, tandis que l'inverse n'est pas vrai. Cependant, il faut garder à l'esprit qu'à un moment donné, nous sommes venus d'un ancêtre commun, donc certains allèles peuvent être des MII. Dans l'analyse PCA ci-dessous, seul le concept IBS est utilisé, bien que dans des analyses plus complexes, il prenne en compte les tests de signification statistique, les limites phénotypiques, la taille des grappes, l'âge et le sexe de la personne, ainsi que des informations supplémentaires sur la structure de la population.
Plus le nombre d'allèles différents est élevé dans deux échantillons, moins ils sont similaires et plus ils sont éloignés l'un de l'autre. La valeur IBS pour ces échantillons sera faible. Mais pour les parents et leurs enfants, l'IBS sera très élevé.
Connaissant les valeurs IBS pour chaque paire d'images dans l'ensemble de données, vous pouvez effectuer une analyse PCA pour voir comment elles sont regroupées.
Le test génétique Atlas utilise un algorithme beaucoup plus sophistiqué pour déterminer la représentation de la population dans les données de génotypage.
Données utilisées
Nous vous rappelons que le manuel utilise des données ouvertes spécialement sélectionnées du projet
1000 Genomes . Pour l'analyse, 10 échantillons contenant des informations sur le génotype d'environ 85 millions de variations ont été sélectionnés, qui ont été obtenus en analysant les données NGS alignées sur la version du génome GRCh37. Les relations familiales et les populations de ces échantillons sont illustrées à la figure 1.
Création de grappes de population
Utilisez les trois fichiers au format Plink obtenus précédemment dans la tâche n ° 1:
CEI.1kg.2019.demo.subset.bed CEI.1kg.2019.demo.subset.bim CEI.1kg.2019.demo.subset.fam
Déterminez la distance par paire entre les 10 échantillons de l'ensemble de données d'apprentissage et tracez une PCA basée sur l'IBS (identité par état). Cela peut être fait comme suit:
Le paramètre
—genome est juste responsable du calcul par paire de l'IBS / IBD entre tous les échantillons dans l'ensemble de données. Le paramètre «
—read-genome » est la matrice de distance par paire obtenue précédemment, et les paramètres «
—cluster —mds-plot 10 sont responsables de l'analyse de l'ACP et de la sortie de ses résultats dans le tableau des 10 premiers composants majeurs. En fait, ce sont les coordonnées de chaque échantillon dans un espace à 10 dimensions.
La dernière commande va créer 4 fichiers dans le dossier:
CEI.1kg.2019.demo.subset.clustering.cluster1 CEI.1kg.2019.demo.subset.clustering.cluster2 CEI.1kg.2019.demo.subset.clustering.cluster3 CEI.1kg.2019.demo.subset.clustering.mds
Nous aurons besoin des deux derniers fichiers de la liste.
La figure 2 montre à quoi ressemble le fichier reçu sur l'ensemble de données de formation MDS. Les champs FID (Family ID) et IID (Individual ID) correspondent aux identificateurs de famille et d'échantillon individuel. Les champs C1 à C10 contiennent les valeurs de chacun des dix composants principaux pour chaque échantillon, où le composant C1 explique au maximum la variabilité des données génétiques des échantillons analysés et C10 au minimum.
 Figure 2
Figure 2 Fichier MDS avec des valeurs de 10 composants principaux pour chaque échantillon.
Lors de la construction d'un diagramme de dispersion à l'aide de deux composants (dans un espace à deux dimensions), on peut détecter des grappes correspondant à la population de l'échantillon. La figure 3 montre des diagrammes de dispersion pour les paires de composants principaux C1xC2, C2xC3 et C1xC3. Lorsque l'on compare les grappes obtenues avec l'affiliation de population de référence (figure 1), la paire des deux premières composantes C1 - C2 montre la plus grande précision (100%), séparant correctement tous les échantillons en fonction de leur affiliation de population déclarée dans le projet 1000 Génomes. Cependant, il est toujours judicieux de comparer les résultats obtenus pour plusieurs paires de composants en raison du chevauchement ou de la séparation possible de clusters réels.
 Figure 3
Figure 3 Diagrammes de dispersion des emplacements d'échantillons pour les paires de composants principaux; l'emplacement des marqueurs a été légèrement modifié pour éviter leur chevauchement.
La création de diagrammes 3D à l'aide des trois premiers composants principaux peut également aider à déterminer le clustering, mais pas toujours. Par exemple, la construction d'un tel diagramme pour les données de la figure 3 nous permet d'identifier 4 grappes dans lesquelles les échantillons des populations PUR et KHV sont regroupés selon la population, et les échantillons de la population CDX sont divisés en deux grappes (figure 4). Ceci est également visible sur la figure 3 dans les coordonnées C2xC3 et C1xC3.
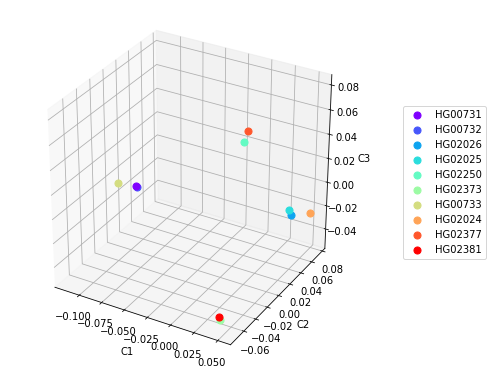 Figure 4
Figure 4 Diagrammes de dispersion pour les trois principaux composants.
Ces résultats contradictoires de l'analyse peuvent s'expliquer par un petit nombre d'échantillons, car les valeurs des principaux composants de chaque échantillon sont différentes pour différents ensembles de données en taille et en composition, et lorsque des échantillons supplémentaires de différentes populations sont inclus, le résultat du regroupement peut changer. Des erreurs sont également potentiellement possibles lors de la création d'un ensemble de données et de la fourniture de données de référence sur la population d'échantillons, cependant, dans le projet 1000 Génomes, la probabilité d'une telle situation est assez faible.
Un fichier MDS n'utilise pas de tabulations ou de virgules comme délimiteurs, ajustez donc son format pour plus de commodité. Utilisez
tab ou
csv comme deuxième argument:
L'équipe va créer le fichier
CEI.1kg.2019.demo.subset.clustering.mds.tab , que vous pouvez télécharger et créer des diagrammes de dispersion similaires à ceux illustrés à la figure 3. Comparez les résultats, ils doivent être identiques à ceux indiqués ci-dessus.
Construire un arbre de clustering
Vous pouvez en outre évaluer le regroupement d'échantillons à l'aide d'un arbre binaire, qui représente les informations de regroupement sur les échantillons sous une forme discrète. Les informations sur cet arbre sont contenues dans le fichier
CEI.1kg.2019.demo.subset.clustering.cluster3 (figure 5).
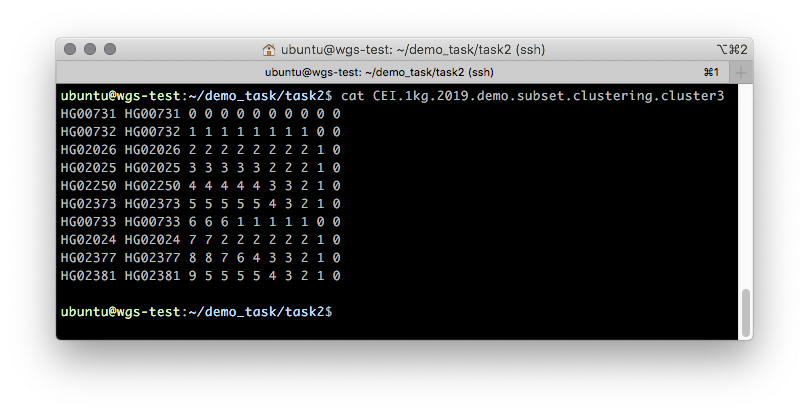 Figure 5
Figure 5 .cluster3 approximatif d'un fichier
.cluster3 qui décrit le processus de regroupement pas à pas d'échantillons de 1 cluster vers N, où N est le nombre d'échantillons.
Les deux premières colonnes de ce fichier contiennent le FID et l'IID. L'affiliation à un cluster est décrite par tous les autres. Ce fichier doit être lu de droite à gauche dans des colonnes par incréments d'une colonne: initialement, tous les échantillons appartiennent à un cluster «0» - la colonne la plus à droite. Lorsqu'il est divisé en deux grappes (dans la deuxième étape, dans la deuxième colonne), deux grappes apparaissent: «0» et «1», où la grappe «0» contient des échantillons HG00731, HG00732 et HG00733 et la grappe «1» contient tout le reste. Une illustration d'une telle partition est présentée à la figure 6.
À partir de l'arbre, on peut conclure que les échantillons appartiennent à la population (figure 1). De plus, la construction de cet arbre nous permet d'établir la proximité des populations individuelles, à savoir, l'occurrence des populations CDX et KHV dans une superpopulation EAS (déjà à la première étape de la division des superpopulations EAS et AMR sont séparées en deux branches existantes). De plus, la construction d'un arbre de regroupement peut aider à corriger les résultats ambigus de la visualisation d'échantillons sur les principaux composants.
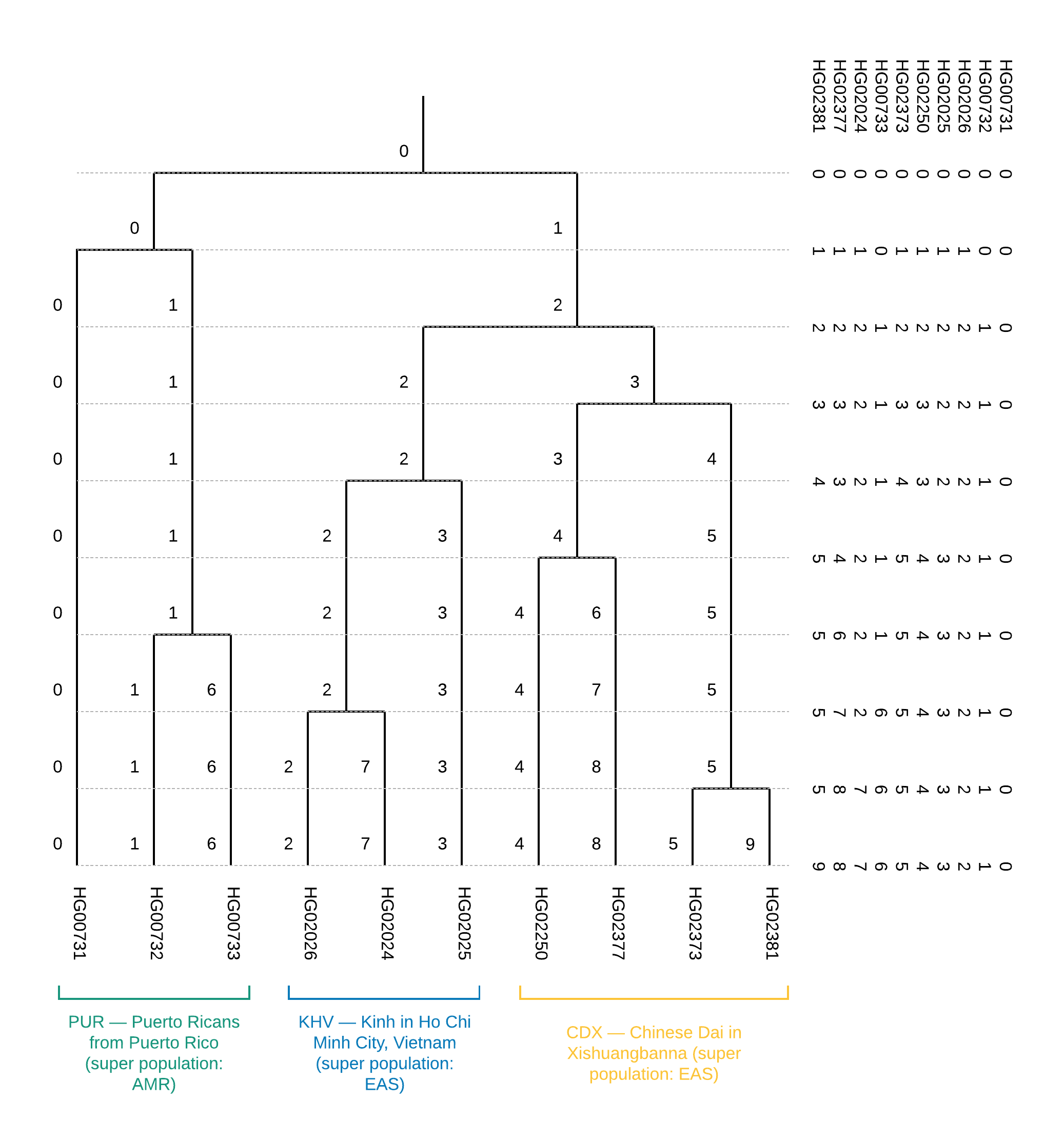 Figure 6
Figure 6 Un arbre de regroupement binaire pour un ensemble de données d'apprentissage de 10 échantillons: à droite se trouve le contenu du fichier
CEI.1kg.2019.demo.subset.clustering.cluster3 (de droite à gauche dans le fichier, de façon identique de haut en bas sur la figure).
La deuxième tâche du concours
Utilisez l'ensemble de données de test à partir de 12 échantillons
Data/Test/CEI.1kg.2019.test.vcf.gz et l'exemple ci-dessus (figure 5) pour créer une arborescence de clustering binaire à partir du fichier
.cluster3 que vous avez
.cluster3 et le joindre à la solution. Analysez l'arbre résultant et tirez des conclusions sur le nombre de superpopulations présentées dans l'ensemble de données de test.
Déterminer le regroupement de la population de 12 échantillons de l'ensemble de données de test en analysant les principaux composants C1, C2 et C3 en tenant compte de l'arbre construit et l'indiquer sur le pedigree construit dans le problème n ° 1, en restreignant les blocs de population individuels (similaire à la figure 1). Les échantillons qui n'ont pas montré la présence de parenté dans le problème n ° 1 doivent être placés de la même manière à l'intérieur des blocs obtenus dans le diagramme sans les relier avec des lignes avec d'autres échantillons. N'oubliez pas de joindre les diagrammes de dispersion que vous avez construits.
Les réponses
doivent être envoyées à
wgs@atlas.ru mail jusqu'au 26 décembre à 23h59. Une autre tâche sera publiée prochainement et les résultats définitifs de ces tâches seront publiés le 28 décembre. Le gagnant recevra le test du génome complet et les deuxième et troisième places recevront le test génétique Atlas. Il y aura également des prix spéciaux de
Yandex.Cloud . Les anciens et actuels employés d'Atlas ne participent pas au concours;)