Aujourd'hui, nous publions la dernière tâche du cycle dans laquelle nous expliquons comment travailler avec des données génétiques.
Les première et
deuxième tâches sont déjà publiées: elles peuvent être résolues et nous envoyer des réponses. Nous vous avertissons que cette tâche prend plus de temps que les autres.
Le prix principal est le
génome complet .

Nous avons précédemment partagé des informations et des liens utiles qui peuvent être utiles pour travailler avec des données bioinformatiques. Nous vous recommandons de lire d'abord les articles précédents si vous les avez manqués:
Qu'est-ce que le génome complet et pourquoi est-il nécessaireNuméro de tâche 1. Découvrez le sexe et le degré de relation.Numéro de tâche 2. Détermination de la structure de la populationClause de non-responsabilitéLe travail avec les données génétiques est effectué sur les systèmes Unix (Linux, macOS), car certaines commandes et logiciels ne sont pas disponibles sur Windows. Par conséquent, pour les utilisateurs de Windows, l'une des solutions les plus simples consiste à louer une machine virtuelle Linux.
Toutes les opérations décrites ci-dessous sont effectuées sur la ligne de commande - terminal. Avant de commencer, découvrez comment travailler dans un terminal exécutant votre système d'exploitation et utilisez des commandes, car certaines d'entre elles peuvent potentiellement endommager le système d'exploitation et vos données.
Logiciels requis
Nous avons collecté l'
image d'une machine virtuelle (VM) avec tous les logiciels nécessaires sur Yandex.Cloud. Les instructions pour configurer une VM et installer le logiciel se trouvent dans l'
article avec la première tâche. Il y a également une instruction sur la façon de configurer la machine pour l'utiliser gratuitement jusqu'au 31 décembre 2019.
Dans cette tâche, vous devez convertir les données de génotypage du format VCF au format 23andMe, télécharger les fichiers reçus vers le service Promethease et vous familiariser avec le contenu du rapport pour chaque échantillon.
Le format 23andMe est un format texte pour stocker des données de génotypage et contient 4 champs séparés par des tabulations. Le premier champ contient l'identifiant de variation (par exemple, rsID), le second contient le chromosome (les valeurs valides pour ce champ sont 1-22, X, Y et MT), le troisième contient la position sur le chromosome, le quatrième contient le génotype (diploïde en présence de deux chromosomes homologues, haploïde dans d'autres cas). Ce format est pris en charge par de nombreux services d'interprétation, nous allons donc travailler avec lui dans la tâche.
Pour terminer la tâche, vous avez besoin du progiciel BCFtools. Si vous ne l'avez pas encore installé, lisez l'
article avec la première tâche. Il contient des instructions d'installation. Nous vous rappelons que pour participer au concours du Nouvel An 2019, toutes les tâches doivent être terminées.
En plus de BCFtools, vous devrez
create_23andme.sh fichier
create_23andme.sh - un script bash qui est utilisé pour générer des données au format 23andMe. Ce fichier se trouve dans le répertoire
/Technical sur Yandex.Cloud ainsi que dans l'archive à télécharger, disponible via le lien dans l'
article .
Prenez note
Il existe de nombreux services qui analysent les données de génotypage: MyHeritage, Promethease, FamilyTreeDNA, DNA.LAND, GEDmatch. Ils fournissent le téléchargement de données de génotypage dans différents formats, souvent spécifiques à un fournisseur de génotypage particulier (Ancestry, 23andMe, MyHeritage, FamilyTreeDNA, GenesForGood et autres). Le plus fidèle au format de données est Promethease: vous pouvez télécharger les fichiers VCF et 23andMe dans ce service.
Il existe plusieurs problèmes de compatibilité entre les formats et les services:
- Différentes sociétés utilisent différentes versions du génome pour cartographier les variations génétiques.Ce problème est résolu par ce qu'on appelle le survol, lorsque les positions des variations génétiques dans les données source sont remplacées par les positions correspondantes dans une autre version du génome. Par exemple, Atlas fournit des données de génotypage pour la version du génome GRCh38, et GEDmatch reçoit des données pour la version précédente du génome GRCh37. La conversion des coordonnées des variations génétiques de GRCh38 à GRCh37 est appelée l'ascenseur.
- Utiliser des identifiants uniques pour les variations génétiques autres que les rsID. Ces incompatibilités sont résolues en excluant ces entrées du fichier ou en les annotant en attribuant un rsID. La seconde n'est pas toujours possible.
- Les services utilisent un ensemble fixe de variations génétiques. Parfois, une non-concordance d'au moins une partie des données en cours de téléchargement entraîne une erreur de chargement. Ce problème est pertinent, par exemple, pour MyHeritage. Il peut être résolu en mettant en évidence un ensemble d'identifiants de variations génétiques qui ne provoquent pas d'erreur de chargement.
Données utilisées
Nous vous rappelons que ce manuel utilise des données ouvertes spécialement sélectionnées du projet
1000 Genomes . Pour l'analyse, nous avons sélectionné 10 échantillons avec des informations sur le génotype de ~ 85 millions de variations, qui ont été obtenues en analysant les données NGS alignées avec la version du génome GRCh37. Les relations familiales et les populations de ces échantillons sont illustrées à la figure 1.
 Figure 1
Figure 1 Le pedigree des échantillons utilisés dans le VCF (le carré correspond au sexe masculin, le cercle à la femelle). La ligne pointillée correspond à une relation de second ordre indéterminée.
Conversion VCF
Vous trouverez ci-dessous des instructions pour convertir un fichier VCF et télécharger les données reçues vers le service Promethease, qui est récemment devenu gratuit. Nous vous suggérons de vous familiariser avec le rapport Promethease reçu sur l'un des échantillons. Utilisez le fichier VCF filtré par la liste des variantes obtenues dans la
tâche n ° 1 .
La commande de
bcftools query vous permet d'extraire toutes les informations disponibles dans un format spécifié par l'utilisateur après l'indicateur
-f d'un fichier VCF. L'indicateur
-s indique l'identifiant de l'échantillon (
HG00731 ) pour lequel extraire les données. L'indicateur -e est utilisé pour indiquer les critères d'exclusion, dans ce cas
'%ID=="."' Exclut les entrées qui n'ont pas de rsID. La sortie de la
bcftools query est transmise au script
create_23andme.sh , qui convertit les données au format TSV avec 4 colonnes (rsID, chromosome, position, génotype) et les écrit dans un fichier. Vous pouvez télécharger et enregistrer vous-même le script
create_23andme.sh pour travailler avec vos propres données de séquençage complet du génome.
Le script
create_23andme.sh utilise les
create_23andme.sh extraits du fichier VCF pour déterminer le type de variation génétique (variation mononucléotidique de SNV, insertion de INS ou suppression de DEL) et écrire l'identifiant rsID, chromosome, position et alleles dans
stdout selon le type de variation spécifique (A, G, T et C sont des allèles valides pour le type SNV, I et D sont des désignations d'allèles valides pour les types INS et DEL).
Gardez à l'esprit que le processus de conversion prend beaucoup de temps: environ 4 heures par fichier pour un échantillon avec environ 1 million de variations. Les BCFtools d'accès simultané ne sont pas pris en charge.
Allez sur
promethease.com et inscrivez-vous. Cliquez sur le bouton Télécharger les données brutes (figure 2) et téléchargez le fichier
HG00731.subset.23andme.txt . Une fois le téléchargement terminé, cliquez sur le bouton Créer un rapport gratuit et entrez le nom souhaité du rapport qui sera généré en fonction de vos données. Après avoir rédigé le rapport, vous recevrez une notification par e-mail et vous pourrez vous familiariser avec le contenu du rapport. Dans les rapports pour chaque échantillon, trouvez le groupe sanguin déterminé par le système d'interprétation Promethease dans le système AB0 / Rh (facteur Rh - Rh). Vérifiez la conformité de vos résultats avec le tableau 1.
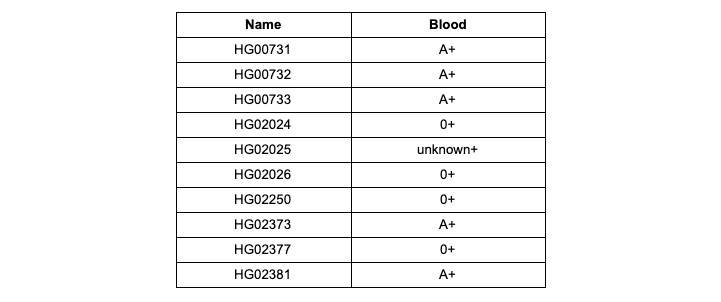 Tableau 1
Tableau 1 . Groupes sanguins et facteur Rh obtenus à partir d'une analyse Promethease d'échantillons d'un ensemble de données de démonstration
L'atlas utilise des seuils qui diffèrent de Promethease pour inclure un attribut particulier dans l'interprétation par niveau de preuve. Le niveau de preuve se réfère à la totalité des résultats des tests statistiques et des critères pour la signification de chaque relation observée entre la variation génétique et toute caractéristique du corps humain. Bon nombre des caractéristiques que l'on peut trouver dans le rapport Promethease ont un faible niveau de preuve et / ou un niveau élevé uniquement dans un ensemble limité de populations, par exemple, uniquement pour les représentants de la population asiatique.
RemarqueEmpiriquement, nous avons installé une liste de variations génétiques basées sur la puce
Infinium Global Screening Array v2.0 qui peut être téléchargée sur MyHeritage. Cette liste (
external_interpretation_rsids.txt ) est stockée dans un fichier séparé dans le répertoire
/Technical , et elle peut être utilisée pour filtrer VCF avec une conversion ultérieure par analogie avec les instructions ci-dessus. Vous pouvez également utiliser ce fichier pour filtrer les données de génotypage à partir d'une puce afin de pouvoir les télécharger sur MyHeritage. Si vous disposez déjà du test génétique Atlas, vous pouvez télécharger les données de génotypage au format depuis votre compte personnel et les filtrer en fonction de la liste de variantes proposée - la première colonne des données téléchargées depuis votre compte personnel.
Notez que les fichiers utilisés dans ce manuel contiennent toujours un champ ALT rempli (allèle alternatif), ce qui permet de comprendre à quel type appartient chaque variation (INS, DEL, SNV) et de créer correctement une entrée au format 23andMe. Les données de séquençage à l'échelle du génome dans l'Atlas contiennent l'allèle ALT rempli uniquement aux endroits où cet allèle a été détecté, sinon les informations pour remplir le champ ALT lors de la génération d'un fichier VCF n'existent tout simplement pas. La production de données sur les sites de référence homozygotes (positions dans le génome où l'allèle de référence n'a pas été trouvé) est nécessaire, car non seulement les variations détectées de la séquence nucléotidique, mais également leur absence ont un effet clinique.
L'absence de l'allèle ALT à de telles positions du génome ne nous permet pas de déterminer le type de variation génétique pour lequel seul l'allèle de référence (REF) a été trouvé. L'enregistrement des génotypes pour de tels cas est compliqué par la nécessité d'utiliser une source d'informations sur les allèles possibles pour cette variation et n'est pas couvert par ce guide. Si vous utilisez potentiellement ce manuel et le script
create_23andme.sh pour convertir un fichier VCF obtenu après un séquençage à l'échelle du génome en Atlas, le fichier converti ne contiendra pas de génotypes homozygotes de référence, car le script
create_23andme.sh filtre explicitement ces enregistrements pour éliminer les erreurs lors de la création d'enregistrements pour les insertions et les suppressions.
Pour que le script
create_23andme.sh produise toujours des génotypes homozygotes de référence, vous devez y remplacer le contenu des lignes 25-28
... if [ "$ALT" == "." ] || [[ "$ALT" == *"*"* ]] then continue fi ...
sur
... if [[ "$ALT" == *"*"* ]] then continue fi if [ "$ALT" == "." ] then echo -e "$RSID\t$CHR\t$POS\t$REF$REF" fi ...
Cette substitution permet d'
stdout entrées
stdout avec des génotypes de référence homozygotes. Il convient de garder à l'esprit que ces entrées pour les insertions et les suppressions seront incorrectes, car les allèles valides dans le format utilisé pour les insertions et les suppressions sont I et D, et le script utilisera les allèles A, G, T ou C. Pour sortir correctement les données pour insertions et suppressions, il est nécessaire de connaître à l'avance quel type de variation est caractéristique d'une position donnée du génome dans laquelle l'allèle ALT n'a pas été détecté. Ces informations peuvent être obtenues en analysant l'allèle ALT s'il est disponible (déjà implémenté dans
create_23andme.sh ) ou en utilisant une base de données externe, par exemple, dbSNP (pas dans
create_23andme.sh ).
Afin d'obtenir un rapport Promethease sur un fichier VCF complet de séquençage complet du génome dans Atlas, vous pouvez charger le fichier VCF lui-même dans Promethease, cependant, vous devez garder à l'esprit que la taille du fichier VCF Atlas compressé est d'environ 8 gigaoctets, tandis que Promethease vous permet de télécharger des fichiers pas plus de 4 gigaoctets. La description des solutions à ce problème est disponible
ici . Une autre solution consiste à diviser le fichier VCF en plusieurs parties (moins de 4 gigaoctets chacune) et à charger chacune en tant que fichier supplémentaire dans le menu de téléchargement des données de Promethease.
La troisième tâche du concours
Téléchargez les données converties de chacun des 12 échantillons du jeu de données de test, que vous avez filtré en fonction de la liste des variations de la première tâche, dans Promethease et compilez un tableau de correspondance pour l'identifiant de l'échantillon - groupe sanguin AB0 / Rh défini par le système d'interprétation Promethease (facteur Rh - Rh). Groupes sanguins identifiés de manière probabiliste et enregistrés avec le préfixe «prob» dans le rapport Promethease, écrivez sans le préfixe. Enregistrer les valeurs indéfinies comme inconnues (le facteur Rhésus pour les groupes sanguins inconnus doit encore être écrit, s'il est défini). Un exemple est présenté dans le tableau 1.
La conversion de VCF au format utilisé ci-dessus dans l'implémentation proposée est grandement simplifiée, mais nécessite un temps considérable. Pour l'optimisation, vous pouvez écrire un script avec une boucle qui générera automatiquement ces données, en itérant sur un ensemble d'identifiants. Il est possible de créer plusieurs scripts de ce type et de transférer chacun différents ensembles d'exemples d'identificateurs pour une exécution parallèle, cependant, le nombre de scripts exécutés en parallèle ne doit pas dépasser le nombre de CPU de votre ordinateur / machine virtuelle. Une bonne description de la création de telles boucles est disponible
ici . Lorsque vous travaillez sur Yandex.Cloud, vous pouvez, si nécessaire, créer une autre machine virtuelle avec un grand nombre de CPU virtuels, ce qui réduira proportionnellement le temps nécessaire pour terminer une tâche.
C'est la dernière tâche de notre cycle. Les réponses
doivent être envoyées à
wgs@atlas.ru mail jusqu'au 26 décembre à 23h59. Nous publierons les bonnes réponses et les noms des gagnants le 28 décembre. Le gagnant recevra le test du génome complet et les deuxième et troisième places recevront le test génétique Atlas. Il y aura également des prix spéciaux de
Yandex.Cloud . Les anciens et actuels employés d'Atlas ne participent pas au concours;)