Présentation
«La brume avant l'aube s'est retirée à contrecœur le long des ravins, des tiges de seigle sont apparues, scintillantes sous le vent. Les oiseaux avaient déjà réussi à se réjouir le matin et bavardaient discrètement par-dessus leur oreille. Les dernières gouttes de sommeil tombèrent dans une tasse de café aromatique. Il est agréable de rencontrer le disque du soleil, d'étirer les articulations givrées et de regarder au loin. C'est qui? La pensée se figea lorsque son regard glissa sur un chemin qui partait de la forêt. Un large sourire illumina son visage. Dès les premiers mouvements, il la reconnut. Elle seule pouvait bouger avec une telle grâce et grâce d'une biche. La main se figea à mi-chemin de la table. Continuant à sourire, il se tourna brusquement brusquement et entra dans la cuisine. Une autre tasse et un plateau de baies sont apparus sur la table. L'odeur du sirop de lavande inondait la véranda. Ce serait une bonne journée, pensa-t-il, un agréable petit déjeuner - c'est sûr.
Une poignée de framboises a rapidement disparu du plateau baie après baie. Une douce voix féminine a annoncé les dernières nouvelles. La semaine dernière, ils n'ont pas mis une seule croix dans le cimetière de la ville. Nous avons survécu! - Une expiration de joie jaillit des poumons déjà raccourcis par l'âge. Oui! »Elle lui répondit. Ces foutus quart de siècle. Un quart de siècle qui n'a laissé aucune trace lisse sur votre visage.
Ses yeux déjà profondément myopes la regardaient et en eux elle voyait les lignes sans fin des graphiques du spectromètre de masse, voyait cette fatigue sans fond qui ne reculait pas dans les tentatives de le mettre dans une boîte. Il a fait face, a résisté. Les gens n'avaient plus une terrible peur animale de périr, probablement la mort la plus terrible qu'on aurait pu imaginer ... "
C'est le début d'une histoire fantastique que je lis à loisir. Il décrit un nouveau type d'arme biologique. Horrible dans son pouvoir destructeur. Les gens ont été stupéfaits lorsqu'ils ont reconnu leur sort. La peur de ce malheur invisible et inévitable était pire que la mort elle-même.
"Prurit"Grande-Bretagne, première moitié du XVIIIe siècle. Le brouillard se dissipe sur les champs verts juteux. Un grand troupeau de moutons se déplaçant lentement vers la rivière. Soudain, nous remarquons quelque chose d'inhabituel. Au moins un cinquième des moutons roule furieusement le long de l'herbe et frotte leur peau sur des pierres qui dépassent de la surface de la terre, laissant des lambeaux en désordre. Peigné, perdu les poils du côté recouverts d'ulcères terribles et d'érosion. Une partie des moutons ne peut plus démanger, ils se contentent lentement, d'une démarche tremblante, de grincer des dents à travers le champ, jusqu'au lieu de la dernière tranquillité. Quel genre de malheur est-ce, pensaient les éleveurs, appelant la maladie selon sa principale manifestation - SCRAPIE ("prurit"). Cette infection n'a pas reculé depuis des siècles, apparaissant de temps en temps ici et là, laissant des familles ruinées.
Vers de cerveauLes vrais scientifiques ne sont pas des gens tout à fait ordinaires, connaissant leurs biographies, on s'étonne souvent de la folie du baril de fortune.
Un des fils de notre histoire débutera par la biographie de Daniel Carlton Gaidusek (1923-2008). Imaginez un jeune homme, il a 23 ans, il vient de recevoir une maîtrise à Harvard, il part avec beaucoup d'enthousiasme travailler à la California Technological University, et pas avec n'importe qui, mais avec Linus Pauling lui-même (deux fois lauréat du prix Nobel). Trois ans plus tard, il accepte l'invitation et prend le poste de chercheur à la faculté de pédiatrie et des maladies infectieuses de son alma mater. Malgré une carrière aussi réussie, quelque chose ne va pas bien et ne lui donne pas la paix. N'ayant pas travaillé depuis 3 ans, il jette tout et part d'abord pour Téhéran à l'Institut Pasteur, et trois ans plus tard, dans un étrange zigzag à travers l'Hindu Kush, il se retrouve au Walter and Eliza Hall Medical Institute de Melbourne. Rétrograder, pas autrement.
C'est en Australie qu'a eu lieu la rencontre fatidique de Daniel Gaidusek avec le médecin-conseil Vincent Zigas (1920-1983), qui a étroitement interagi avec les tribus de Papouasie-Nouvelle-Guinée, leur fournissant des soins médicaux. Zigas parle à Daniel d'une maladie inconnue, dont les symptômes étranges se manifestent dans une seule nation - Foret. Gaidusek se précipite avec impatience pour apprendre la langue des aborigènes et quelques mois plus tard, Zigas apporte et représente Gaidusek du groupe ethnique Foret. Pendant près d'un an, ils vivent au sein d'une tribu sauvage, traquant toutes les habitudes et coutumes. Observez les patients et réalisez l'autopsie des morts. [1]
Voici comment ils décrivent la séquence de développement d'une maladie qui les intéresse dans leur article:
«... L'apathie et la fatigue écrasante envahissent une personne. Après un mois ou un peu plus, des secousses et des tremblements caractéristiques commencent. Le tremblement des membres, du tronc et de la tête devient plus distinct et constant. Une personne perd la capacité de bouger. Entre un et deux ans, la mort survient. Les membres de la tribu Phore appellent cette maladie «Kuru», ce qui signifie trembler, gâter. Et ils croient que la raison réside dans le mauvais œil du chaman. "
Après une autopsie, il a été constaté que Zigas et Gaidusek, décédés de la maladie, avaient transformé le cerveau en une substance spongieuse. [2]
La résidence à long terme au sein de la tribu a permis à Haidusek et Zigas de découvrir la cause de la maladie. Il s'est avéré que la tribu Foret pratiquait le cannibalisme.
Après la mort de l'un des membres les plus âgés du genre, son corps a été sculpté, la boîte du crâne a été ouverte et le cerveau a été mangé, car on croyait que manger le cerveau était un rituel des derniers honneurs pour le défunt, et celui qui mange le cerveau acquiert sa sagesse, son courage et d'autres nobles qualités, dont il était propriétaire. Habituellement, la majeure partie du cerveau était mangée par les femmes et, par conséquent, parmi elles, le nombre de cas était plus élevé [3]. Avec l'éradication d'une coutume aussi pernicieuse, la maladie de Kuru a été presque complètement vaincue.
Pour la description de la maladie de Kuru en 1976, Gaidusek a reçu le prix Nobel. Et ici, les motifs du comité Nobel, qui a retenu l'attention de Vincent Zigas, ne sont pas clairs. Dans sa conférence Nobel, Gaidusek a parlé de la nature virale de la maladie de Kuru. Nous apprenons s'il avait raison ou non plus tard.

Un patient affecté par la "poule".
"J'ai eu le temps"En attendant, avance rapide vers l'Allemagne. Au début du XXe siècle, une clinique psychiatrique à Breslau, un service dirigé par Alois Alzheimer. Un jeune médecin de navire vient travailler, qui a décidé de devenir neurologue. Bien qu'il comprenne constamment les bases de la profession, il parvient à trouver des patients atteints d'une maladie inconnue jusqu'à présent. La recherche est interrompue par la Première Guerre mondiale, qui a renvoyé le Dr Hans-Gerhard Kreutzfeld dans la marine. Ce n'est qu'en 1920, après 6 ans, qu'il publie une description de la maladie.
Dans la description, nous constatons que les patients ont perdu leur mémoire à grande vitesse, ont cessé d'être conscients d'eux-mêmes et sont décédés 8 à 12 mois après les premières manifestations du tableau clinique. Dans les préparations cérébrales obtenues de ces patients, des «structures spongieuses» caractéristiques ont été trouvées.
Il vaut la peine de dire qu'il a été incroyablement chanceux s'il avait six mois de retard, et le vent du temps aurait dissipé son nom pendant des siècles, car quelques mois plus tard, le travail d'Alfons Jacob avec une description de la même maladie qui a trouvé le nom de ses découvreurs - la maladie de Creutzfeldt-Jakob (CJK).
Qu'est-ce qui pourrait être commun entre la tremblante, le kuru et la CJK? C'est la question que les scientifiques ont commencé à se poser dans les années 50 du 20e siècle, car ces maladies étaient tellement similaires à une longue période d'incubation de 5 à 10 ans et au triste sort invariable de la personne affectée, qu'il s'agisse d'un animal ou d'une personne. De plus, les dommages ont principalement atteint le cerveau. Ce groupe de maladies neurodégénératives avec une longue période d'incubation a été appelé.
Corps principal
Les expériences
Avec le développement de méthodes expérimentales de biochimie, il est devenu possible d'approcher enfin ces pathologies. Il était extrêmement difficile de trouver la source de l'infection, à condition que les manifestations de la maladie ne puissent être détectées qu'après des années, malgré les difficultés, les tentatives pour trouver les causes de la maladie ne se sont pas arrêtées. Les laboratoires cherchaient des moyens de simplifier le travail expérimental, en réduisant la période d'incubation à une période acceptable.
Patison et Guiley ont donc pu transmettre la maladie de mouton à mouton en utilisant des filtrats acellulaires. Pour commencer les expériences de laboratoire, il ne restait plus qu'une étape: transférer la maladie du mouton à l'animal de laboratoire. Et Chandler le fait en 1960, dont il écrit un petit mais très célèbre article en 1961 [4]. Il a réussi à infecter une souris de laboratoire avec une substance du cerveau d'un animal malade. De plus, dans des études récentes, la manifestation de la maladie n'a dû attendre que 7 mois. Il est devenu pratique d'étudier la maladie en laboratoire.
La recherche d'un agent infectieux s'est intensifiée. Il n'a pas été possible de l'installer depuis longtemps. Au début, ils ont recherché un virus inconnu, similaire à l'herpès ou à l'encéphalite, mais n'ont rien trouvé. Tous les chercheurs ont été surpris de constater que la capacité d'infecter cette substance, isolée du cerveau d'animaux malades, persistait après un fort réchauffement à long terme et après un traitement à l'acétaminophénylamine. Des expériences ont été réalisées dans lesquelles le filtrat a été traité avec des UV durs et des rayonnements ionisants. Malgré cela, le filtrat a conservé la capacité de s'infecter. [5] Les soupçons ont commencé à se répandre en ce que les virus dans ce cas n'avaient rien à voir avec cela, car les acides nucléiques (un composant indispensable de tout virus) s'effondrent simplement sous un tel impact.
Griffith, dans une courte note sur une page et demie de texte en 1967, a exprimé une pensée hérétique - l'agent infectieux ne contient pas d'acides nucléiques. [6] Il s'agit d'une protéine capable de s'auto-reproduire dans la cellule. C'est avec cette note qu'une nouvelle ère a commencé.
Protéine infectieuse
Les expériences de recherche sur la tremblante sont restées complexes et longues. Seulement 15 ans plus tard, Stanley Pruziner de l'Université de Californie à San Francisco a identifié et décrit un agent qui, sous sa forme pure, peut provoquer le développement de la tremblante. Il s'est avéré que cette substance étonnante est résistante à la chaleur, conserve l'infectiosité après traitement avec divers agents nocifs, tels que: protéinase K, urée, chlorure de guanidine, détergents, SDS et nucléases - enzymes endommageant l'ADN, mais il a également été constaté que cet agent infectieux est sensible aux ionisations rayonnement en présence d'oxygène, caractéristique des protéines hydrophobes ayant une forte affinité pour les lipides. [8]
Pruziner a inventé le nom d'un agent responsable de la tremblante - «PRION» (prion - particule infectieuse protéinacée). La protéine prion (Prione Protein PrP) a été isolée un peu plus tard. Les méthodes de séquençage à cette époque étaient déjà assez bien développées et ont rapidement permis d'établir la séquence PrP primaire. Tout le monde a commencé à chercher la source de la PrP. Un article dans Nature de 1985, qui a marqué la fin de la recherche, a perplexe de nombreux chercheurs: l'ARN matriciel (une molécule - un modèle par lequel les protéines sont ensuite synthétisées) nécessaires à la synthèse de la PrP a été trouvé dans un cerveau sain. [7]
Cela ne signifiait qu'une chose - la protéine responsable du développement de la maladie est toujours présente dans le cerveau, quel que soit le développement de la maladie. Il a été découvert plus tard que le gène codant pour la PrP est présent chez tous les mammifères, ainsi que chez les oiseaux et les poissons.
L'emplacement des sections individuelles du 20e chromosome d'une personne avec une marque de l'emplacement du gène codant pour la PrPC.
Structure des protéines
Quelle est cette incroyable protéine? Sa fonction et après 37 ans à partir du moment de la découverte n'a pas été clarifiée (ici, il convient de le dire grâce au modèle occidental de la science des articles subventionnés). Cette protéine est connue pour être liée à la membrane cellulaire. Et peut-être responsable des interactions intercellulaires dans le cerveau.
Pour comprendre comment une protéine ordinaire devient contagieuse, vous devez examiner la structure des protéines. La structure principale d'une protéine est la séquence de résidus d'acides aminés. Cette séquence est la même dans la PrPC normale et sous la forme infectieuse de PrPSc.
Des différences ont été constatées au niveau de la structure spatiale secondaire et tertiaire. En PrPC, la structure secondaire est représentée par 42% des hélices α et 3% des structures β, tandis que PrPSc contient en même temps 30% des hélices α et 43% des structures β. Ce fait suggère que la forme pathologique de la protéine se forme lors d'un repliement incorrect de la séquence d'acides aminés en couches plissées β.
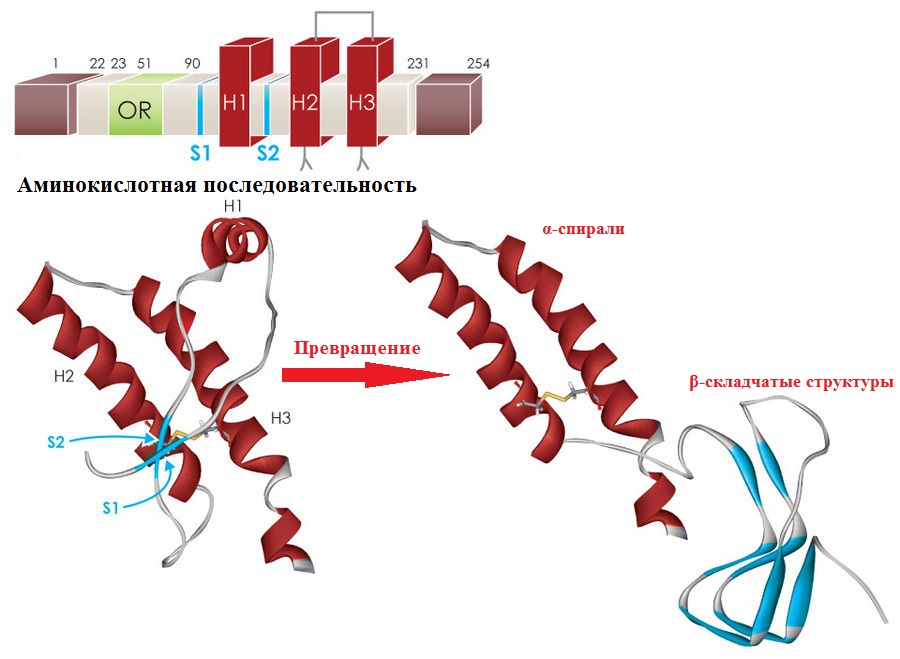
Dans l'image ci-dessus, la séquence d'acides aminés de PrP, avec des régions en surbrillance de diverses structures protéiques des hélices H1, H2, P3 - α. La transformation des spirales en couches pliées β est illustrée ci-dessous. Image: Par Olivia May, Ph.D
Hypothèse du prion
Sur la base des données accumulées en 1991, Pruziner forme l'hypothèse du prion, qui postule ce qui suit:
- l'agent infectieux est la protéine PrPSc,
- l'agent infectieux PrPSc peut se répliquer en l'absence d'acide nucléique,
- la conversion des protéines de la forme normale (PrPC) en infectieuses (PrPSc) se fait par une transition conformationnelle,
- la transition conformationnelle de la PrPC en PrPSc peut se produire spontanément, conduisant à des formes sporadiques de maladies à prions. Elle peut être causée par l'ingestion de la forme pathologique de PrPSc de l'extérieur (formes acquises de maladies à prions),
- la transition peut se produire en raison de mutations du gène Prnp qui contribuent à la formation de PrPSc à partir de PrPC (formes héréditaires de maladies à prions). [9,10].
Ainsi, les maladies à prions peuvent être causées par un défaut génétique, une infection externe ou une combinaison de ceux-ci.
Malgré les ardentes attaques des critiques de cette théorie, maintenant presque tout le monde convient que Pruziner avait raison, et il existe une quantité considérable de preuves expérimentales pour cela. Par exemple, si nous imaginons que la reproduction de la PrPSc après ingestion se produit par le transfert de la conformation pathologique à la PrPC, alors les organismes dépourvus de PrPC devraient être résistants à l'infection à prions. Une telle expérience a été réalisée en utilisant des souris transgéniques homozygotes pour une délétion du gène Prnp (Prnp0 / 0). L'introduction du tissu cérébral frotté de souris atteintes de tremblante à des souris transgéniques Prnp0 / 0 n'a pas conduit au développement de la maladie en raison du manque de PrP normale. De plus, il s'est avéré qu'en l'absence de PrPC non seulement la reproduction des prions, mais aussi les dommages aux tissus nerveux ne se produisent pas.
La preuve finale du concept de prions a longtemps été limitée par l'impossibilité d'obtenir une quantité importante de PrPres, une forme de PrPSc formée in vitro qui résiste à la protéolyse partielle et peut provoquer des maladies lorsqu'elle est administrée à des animaux de laboratoire. Récemment, il a été montré qu'un fragment de PrP de souris recombinant synthétisé dans Escherichia coli forme des fibrilles in vitro qui, lorsqu'il est introduit dans des souris transgéniques exprimant le même fragment de PrP, conduit au développement d'une maladie à prions. [13]
Récemment, un système d'amplification cyclique de la forme prion de la protéine PrP a été développé, à l'aide duquel il est possible de former une quantité importante de PrPres (version pathologique artificielle du prion) in vitro. Cela a permis d'obtenir et de démontrer l'infectiosité du prion synthétisé artificiellement.
Un lecteur attentif remarquera qu'il est peu probable que les prions formés dans le cerveau d'un mouton soient pathogènes pour l'homme. Et ils auront presque raison. Il est connu que la transmission d'une infection à prions entre espèces de mammifères est limitée par des barrières interspécifiques. Par exemple, la maladie de Creutzfeldt-Jakob est transmise d'une personne à l'autre et de l'homme aux chimpanzés; la tremblante est transmise aux ovins et caprins, mais pas aux chimpanzés. En même temps, les barrières interspécifiques ne sont pas absolues. Les barrières interspécifiques peuvent s'exprimer non pas tant par l'impossibilité de transmettre l'infection aux animaux d'une espèce éloignée, mais par l'allongement de la période d'incubation, ainsi que par le fait que tous, mais une partie des animaux infectés expérimentalement deviennent malades. On pense que les barrières interspécifiques sont causées par des différences dans la structure primaire de la PrP et des modifications chez les mammifères de différentes espèces. Cela a été confirmé par les observations suivantes. Les souris transgéniques exprimant la PrP du hamster étaient très sensibles à l'infection par le prion du hamster, contrairement aux souris de type sauvage. La transmission de la maladie de Creutzfeldt-Jakob de l'homme à la souris est limitée par la barrière interspécifique, mais les souris transgéniques exprimant la PrP humaine sont sensibles à l'infection.
Il existe également des difficultés pour infecter les animaux avec de la protéine prion pure. Ces difficultés s'expliquent facilement.
La première raison est que dans les cellules d'un organisme ordinaire, les protéines subissent une modification post-traductionnelle, qui est difficile à reproduire dans des conditions expérimentales.
La deuxième raison est que le prion est une protéine membranaire et il faut supposer que sa structure est plus stable dans les conditions d'un environnement membranaire, comme l'ont montré des études récentes.
Il a été montré chez eux que les prions en présence de cholestérol et de phosphatidyléthanolamine formaient la forme pathogène beaucoup plus facilement et avaient une infectiosité beaucoup plus grande.
Prions - armes biologiques
Sur ce point, on pourrait finir de parler de prions effrayants. Cependant, le lecteur demandera légitimement: "Et qu'en est-il des armes biologiques?" Après tout, pour qu'une infection se produise, il est nécessaire que la molécule de prion pathogène pénètre dans le cerveau. Nous ne ferons pas de craniotomie pour nous-mêmes. »
En fait, la situation avec des voies d'infection possibles s'est révélée bien pire qu'on ne pouvait l'imaginer.
En 1974, le premier cas de maladie iatrogène (due à une exposition externe) de la maladie de Creutzfeldt-Jakob, généralement considérée comme une pathologie génétique, a été décrit.Il existe des descriptions de 3 cas de transmission de la MCJ à la suite d'une transfusion sanguine d'un donneur qui a été diagnostiqué avec la MCJ lors d'une épidémie de la maladie au Royaume-Uni [28]. D'où vient cette épidémie ... Comme d'habitude à cause de la cupidité. La MCJ s'est développée chez l'homme après avoir mangé du bœuf infecté de prions.En 1986, une épidémie de maladie à prions chez les vaches a éclaté en Grande-Bretagne, également appelée «maladie de la vache folle», qui a entraîné la mort de plus de 160 000 bovins [29]. La raison en était l'utilisation de suppléments nutritionnels à base de farine de viande et d'os lorsque, en raison de règles mal contrôlées pour le traitement des sous-produits animaux, la PrPSc provenant de moutons infectés par la tremblante et d'autres bovins est tombée dans l'alimentation des vaches. Habituellement, la technologie pour produire une telle farine après un broyage soigneux de la matière première comprend un traitement avec des solvants gras actifs, ainsi qu'un traitement thermique à une température de 130 ° C. Cependant, à la fin des années 70, les entrepreneurs, ayant décidé d'augmenter la valeur nutritive de la farine de viande et d'os, ont réduit le mode de traitement thermique à 110 ° C et ont également réduit la quantité de substances extrayant les graisses.Ce sont ces changements qui ont contribué à l'émergence et au développement de l'épidémie chez les bovins.Il a été prouvé que l'épidémie chez les vaches a conduit à l'émergence d'un nouveau type de MCJ, appelé "variante CJD" [15]. Les premiers cas de FBC ont été signalés en 1995, lorsque la maladie a été diagnostiquée chez 2 adolescents britanniques [16.17]. En raison de la longue période d'incubation, le lien entre la maladie et la viande infectée au Royaume-Uni n'a été établi que lorsque l'incidence des vaches est devenue une épidémie. L'épidémie a été maîtrisée après un massacre massif de bovins et des changements dans la technologie de production qui ont considérablement réduit la contamination de la viande par des composants du tissu nerveux. Au Royaume-Uni, le nombre annuel de nouveaux cas de BCB, qui a culminé en 2000, diminue régulièrement, et un seul cas a été confirmé en 2013 [18].Chez tous les patients, la MCJ s'est développée après avoir mangé de la viande provenant de bovins malades. Mais, malgré l'épidémie généralisée qui a frappé des centaines de milliers de bovins, relativement peu de personnes qui ont mangé de la viande d'animaux malades ont développé la MCJ [33]. (rappelez-vous la barrière interspécifique).La période d'incubation (le temps entre la consommation de bœuf infecté et la manifestation des symptômes) a été longue: la plupart des patients ont été infectés à la fin des années 80 et l'incidence maximale s'est produite au début des années 2000, c'est-à-dire que la période d'incubation était de 11 à 12 ans. Dans les derniers cas diagnostiqués, la période d'incubation est passée de 12 à plus de 20 ans [18.19].Les manifestations cliniques de la variante de la MCJ sont différentes des autres formes de MCJ. La maladie rattrape les jeunes jusqu'à 30 ans, son apparition se caractérise par des changements de personnalité: le patient perd ses anciens intérêts, commence à fuir les proches, il développe de l'anxiété, de l'insomnie, de la dépression. Les troubles du mouvement surviennent environ six mois après le début de la maladie. La démence survient plus tard que sous la forme classique, le patient est conscient de son aggravation. Assez rapidement, il perd la capacité de libre-service. Non seulement l'apparition à un âge plus jeune, mais aussi le taux de survie moyen dépassant 14 mois est typique pour le cJDV [18,19]. Il est probable que les différences de survie entre la MCJ classique et sa variante soient en partie liées au jeune âge des patients.Ainsi, la nature elle-même nous a démontré la possibilité d'utiliser des prions comme armes avec un temps d'exposition retardé.À mon plus grand regret, en 2011, lors des travaux expérimentaux sur l'étude de la maladie de Creutzfeldt-Jakob, la possibilité d'une infection aéroportée par des aérosols contenant des particules de prion a été montrée chez la souris.Les prions sont-ils une arme biologique idéale?
Quels sont les principaux avantages:- La maladie se manifeste dans un délai, l'attaquant a le temps d'infecter le plus de personnes possible. Dans ce cas, tous ceux qui l'entourent seront dans l'ignorance totale.
- Vous pouvez arrêter l'infection aussi discrètement que vous la démarrez. Trouver des traces et la source de l'infection après 5-7 ans sera incroyablement difficile. De plus, vous devrez savoir quoi rechercher.
- . ,
- .
- , . . , .
- .
- . , [9]
Les prions sont la technologie parfaite pour la terreur. Il existe une technologie bien décrite pour la synthèse de formes pathogènes de protéines prions [13].Même s'il serait difficile pour un terroriste ordinaire d'organiser un laboratoire biochimique, personne ne prend la peine d'utiliser d'innombrables troupeaux d'animaux pour obtenir une grande quantité de substance cérébrale infectée par la MCJ.Personne et rien n'empêcheront les terroristes de commencer la synthèse de masse des protéines prions et de les ajouter au lait en poudre, aux préparations pour nourrissons, à la viande hachée, aux abats de viande et d'os, aux farines de soja ou à toute autre substance dont l'usine de production sera à leur portée.Si nous imaginons qu'un biochimiste talentueux tombera entre les mains de terroristes par la force ou pour des raisons financières, idéologiques et autres, alors personne ne l'empêchera de synthétiser un aérosol lipidique-protéique avec des particules de prion. Ensuite, vaporisez un aérosol indétectable dans les systèmes de ventilation. Cette méthode est plus terrible que par la nourriture, car le nœud nasopharyngé a un lien étroit avec le cerveau et la probabilité d'infection augmente plusieurs fois.Imaginez les effets de l'infection. Après 3-7, ou peut-être tous les 15 ans dans un territoire illimité, le développement de masse de la maladie à prions du cerveau commence. Panique, horreur, peur, destruction. Des villes entières sont des zombies, dont le cerveau se transforme littéralement en éponge. Il n'y a pas de remède, pas d'espoir, seulement l'horreur d'une mort imminente inévitable.Conclusion
L'utilisation de telles armes n'est qu'une question de temps. Par conséquent, vous devez maintenant prendre un certain nombre d'étapes:- Mener des recherches sur la création de systèmes de détection fiables pour les prions pathologiques dans les aliments, l'eau, l'air et rendre ce test obligatoire pour une utilisation dans le monde entier. Introduire des systèmes de détection des protéines prions.
- Rechercher la possibilité de diagnostiquer l'apparition de prions pathologiques chez l'homme. Il y a de bonnes nouvelles qu'une méthode très sensible pour détecter les prions pathologiques a été développée. [12]
- Cherchez un moyen de guérir une personne. Ce qui semble incroyablement difficile, malgré des avancées prometteuses avec des anticorps anti-prion qui peuvent traverser la barrière hémato-encéphalique.
- Suivez les cas massifs de maladies à prions chez les animaux au niveau des services spéciaux.
Souhait du nouvel an
Je souhaite ne jamais rencontrer une seule molécule de PrPSC!Je vais chercher une opportunité pour obtenir un test de diagnostic très sensible - un système pour déterminer si nous avons déjà réussi à nous infecter ...Références aux sources1. www.nobelprize.org/prizes/medicine/1976/gajdusek/biographical2. Gajdusek, DC; Zigas, V. (1957-11-14). "Maladie dégénérative du système nerveux central en Nouvelle-Guinée." Journal de médecine de la Nouvelle-Angleterre. 257 (20): 974–978.3. Hussain Khan, CG Bio-medical Paradigm // Problèmes bio-sociaux de la santé. Rédacteur en chef, RK Pathak. New Delhi: Northern Book Center, 2008. - p. 15
4. Chandler RL (1961) Lancet, 1,1378–1379
5. Alper T., Cramp WA, Haig DA, and Clarke MC (1967) Nature, 214, 764–766.
6. Griffith JS (1967) Nature, 215,1043–1044.
7. Chesebro B., Race R., Wehrly K., Nishio J., Bloom M., Lechner D., Bergstrom S., Robbins K., Mayer L., Keith JM, et al. (1985) Nature,315, 331–333.
8. Prusiner SB (1982) Science, 216,136–144
9. Prusiner SB (1991) Science, 252,1515–1522
10. Prusiner SB (1993) Proc. Natl. Acad. Sci. USA Vol. 90, pp. 10962-10966, December 1993 Biochemistry
11. Saima Zafar et al., Handbook of Clinical Neurology, Vol. 165, 2019 (3rd series)
12. Serena Singh, Mari L. DeMarco JALM, January 2020
13. Nature Communications (2018) Chae Kim, Xiangzhu Xiao, Shugui Chen, Tracy Haldiman, Vitautas Smirnovas, Diane Kofskey, Miriam Warren, Krystyna Surewicz, Nicholas R. Maurer, Qingzhong Kong, Witold Surewicz & Jiri G. Safar Artificial strain of human prions created in vitro volume 9, Article number: 2166
14. Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG. Possible transmission of variant Creutzfeldta Jakob disease by blood transfusion. Lancet. 2004;363:417-421.
15. Collinge J. Human prion diseases and bovine spongiform encephalopathy (BSE). Hum Mol Genet. 1997;6(10):1699-1705
16. Bateman D, Hilton D, Love S, Zeidler M, Beck J, Collinge J. Sporadic Creutzfeldt Jakob disease in a 18-year-old in the UK. Lancet. 1995; 346(8983):1155-1156.
17. Britton TC, al-Sarraj S, Shaw C, Campbell T, Collinge J. Sporadic Creutzfeldt—Jakob disease in a 16-year-old in the UK. Lancet. 1995; 346(8983): 1155.
18. Soomro S, Mohan Ch. Biomarkers for sporadic Creutzfeldt Jakob disease. Annals of Clinical and Translational Neurology. 2016;3(6):465-472.
19. Imran M, Mahmood S. An overview of human prion diseases. Virol J. 2011;8:559.