Analisis afiliasi populasi manusia oleh DNA, dalam pengalaman kami, menimbulkan tiga pertanyaan besar di masyarakat: dapatkah gen dan kelompok etnis dihubungkan bersama, bagaimana analisis asal dari sudut pandang teknis, dan apakah tes genetik dapat "mengidentifikasi orang Yahudi". Untuk beberapa alasan, justru masalah identitas Yahudi dengan DNA yang mengkhawatirkan baik mereka yang memiliki bukti tak terbantahkan milik orang-orang pilihan Tuhan dan mereka yang tidak makan matzo dan tidak membaca Taurat.
Dalam materi Genotek baru di Geektimes, kami akan mencoba menjawab semuanya secara berurutan. Dan ya, kami juga mendefinisikan Yahudi.

Ras alias kelompok populasi dalam biologi, kedokteran dan genetika
Manusia memiliki kebiasaan buruk membenarkan kekerasan dengan superioritas "bawaan" dari satu ras ke ras lain - inilah mengapa ahli biologi modern mendekati isu perbedaan genetis antara populasi dengan kehati-hatian pencari ranjau. Keberadaan (non) batas biologis antara kelompok ras dan etnis telah dibahas dengan serius sepanjang abad ke-20, tetapi belum ada konsensus akhir yang dicapai tentang masalah ini (
1 ).
Diharapkan bahwa urutan genom manusia akan menyatukan semua orang. Genom, baca "dari" dan "ke", akan menunjukkan bahwa batas antara kelompok bersifat sosial, dan gen itu sama untuk semua orang. Ternyata berbeda: sebuah studi hati-hati dari kode nukleotida manusia dihidupkan kembali dan meningkatnya minat pada perbedaan biologis antara ras dan populasi etnis. Gen yang sama, secara umum, menemukan varian alelik yang sedikit berbeda terkait dengan risiko penyakit (
2 ), metabolisme obat (
3 ), respons tubuh terhadap kondisi lingkungan (
4 ), dan varian ini ditemukan pada populasi yang berbeda dengan frekuensi yang berbeda.
Pencarian untuk gen "India" atau "Afrika" yang tidak ada telah dihentikan, tetapi penelitian di bidang genetika medis dan populasi masih menarik paralel antara karakteristik biologis dan etnis peserta. Penggunaan istilah "ras" dan "etnis" dalam karya-karya tersebut dibahas secara aktif (dan sering dikutuk). Ada upaya untuk memperkenalkan aturan yang memaksa peneliti untuk membenarkan kebutuhan untuk menggunakan kategori "licin" dan untuk memperjelas apa yang dimaksud dengan istilah tertentu. Pada bulan Februari tahun lalu, Science, salah satu jurnal sains yang paling dihormati, menerbitkan sebuah artikel yang ambigu (
5 ), mengusulkan untuk sepenuhnya mengabaikan penggunaan istilah "ras" dalam penelitian genetika, menggantikannya dengan "leluhur" yang lebih benar dan netral - "asal" .
Tetapi bahkan dalam kondisi ketidakpastian dengan persyaratan, umat manusia masih dibagi menjadi kelompok populasi: khususnya, untuk melakukan uji klinis obat yang benar dan penilaian risiko penyakit. Sebagai contoh, tiga varian alelik dari gen NOD2 - R702W, G908R dan 1007fs - dikaitkan dengan peningkatan risiko penyakit Crohn di Eropa Amerika (
6 ,
7 ), namun, tidak satupun dari varian ini terkait dengan penyakit Crohn di Jepang (
8 ). Alel gen CCR5 diketahui mempengaruhi tingkat pengembangan immunodefisiensi pada pasien yang terinfeksi HIV (
9 ): di antara mereka, sebuah pilihan ditemukan yang memperlambat perkembangan penyakit pada orang Amerika keturunan Eropa, tetapi mempercepat perkembangannya di Afrika Amerika (
10 ). Orang Asia menemukan korelasi antara polimorfisme gen protein p53, yang mengatur respons stres dan menekan perkembangan tumor, dan suhu musim dingin rata-rata di habitat populasi - adaptasi genetik terhadap salju (
11 ). Dan jika di masa lalu hanya informasi yang disediakan oleh para peserta itu sendiri yang digunakan untuk memecah sampel menjadi kelompok etnis, di era pasca-genom mereka semakin dilengkapi dan disempurnakan dengan penilaian genetik dari asal subjek.
Variasi genetik antar populasi
Dalam kehidupan sehari-hari, kami membagi orang ke dalam kelompok sesuai dengan penampilan atau bahasa komunikasi. Kebanyakan orang Denmark lebih mirip satu sama lain daripada mereka masing-masing menyerupai orang Italia (di
sini adalah visualisasi keren dengan potret rata-rata dari berbagai negara). Orang-orang Denmark dan Italia jauh lebih dekat satu sama lain daripada mereka masing-masing - dengan penduduk Afrika sub-Sahara: fenotipe manusia dikelompokkan menurut pola geografis. Distribusi genotipe memiliki struktur yang serupa: anggota kelompok lokal, sebagai aturan, memiliki ikatan keluarga yang lebih dekat daripada penduduk di daerah terpencil, dan populasi yang tinggal di satu wilayah lebih dekat daripada mereka yang habitatnya dipisahkan oleh hambatan geografis (misalnya, pegunungan atau air array).
Selain itu, keragaman genetik populasi manusia lebih rendah daripada spesies biologis banyak. Ini dijelaskan oleh fakta bahwa manusia adalah spesies muda: kelompok individu memiliki waktu yang relatif sedikit untuk mengakumulasi perbedaan. Dua orang yang dipilih secara acak berbeda satu sama lain dengan masing-masing ~ 1000 nukleotida, sedangkan dua simpanse tidak bertepatan sekali dalam ~ 500 "huruf". Namun, secara total, ada sekitar 3 juta "titik perbedaan" potensial dalam genom manusia. Sebagian besar perbedaan ini, yang disebut polimorfisme nukleotida tunggal (SNP), bersifat netral atau hampir netral, tetapi beberapa di antaranya bertanggung jawab atas perbedaan fenotipik antara manusia.
Distribusi polimorfisme netral (karena tidak memiliki makna biologis, mereka tidak dikenai seleksi evolusioner terarah, mereka dibawa oleh angin migrasi) pada populasi dunia mencerminkan sejarah demografis spesies kita. Bukti genetik dan arkeologis menunjukkan bahwa selama 100.000 tahun terakhir ukuran populasi manusia telah tumbuh secara signifikan. Orang-orang menetap di luar Afrika, menjajah seluruh dunia. Proses pemukiman kembali memengaruhi distribusi geografis alel dalam dua cara: pertama, "efek pendiri" mempengaruhi - dalam populasi imigran, sebagai aturan, hanya sebagian varian genetik dari seluruh kumpulan keragaman mereka dalam populasi leluhur yang diwakili; kedua, apa yang disebut "assortative crossing" terjadi, yaitu pasangan-pasangan terbentuk terutama dalam kelompok mereka, yang membatasi distribusi polimorfisme de novo yang ada dan muncul di antara individu-individu yang menghuni wilayah geografis yang berbeda. Proses-proses ini menyebabkan akumulasi bertahap dari perbedaan genetik.
Dalam konteks kelompok populasi, penanda genomik mulai dipelajari pada tahun 70an - 80an, pada tahun 90an mereka mulai digunakan untuk mengidentifikasi populasi orang tertentu. Para peneliti telah berulang kali menunjukkan bahwa polimorfisme genetik dapat berhasil mengisolasi kelompok populasi dan menentukan afiliasi kelompok seseorang. Kemudian ditunjukkan bahwa orang yang hidup di benua yang sama biasanya lebih dekat satu sama lain secara genetik daripada orang-orang dari benua yang berbeda. Awalnya, dalam studi tersebut, informasi tentang tempat lahir, ras, kelompok etnis sudah diketahui sejak awal dan digunakan bersama dengan data genetik; Jika subyek didistribusikan secara membabi buta di antara kelompok-kelompok semata-mata berdasarkan sifat-sifat genetik, korespondensi antara asal geografis, etnis, dan struktur populasi kurang jelas. Seperti yang ditunjukkan oleh penelitian lebih lanjut, keberhasilan bergantung pada penanda genetik yang digunakan dan jumlahnya (lebih banyak lebih baik), pilihan populasi referensi yang tepat dan faktor-faktor lain (
12 ).
Pada 2004, di Amerika Serikat, definisi genetik populasi tidak hanya digunakan dalam penelitian biomedis, tetapi juga dalam penyelidikan kejahatan: artikel dari Nature ini memuat kisah menarik tentang bagaimana para petugas kepolisian, yang sangat ingin menemukan seorang penjahat, memerintahkan tes DNA dari sebuah perusahaan komersial, memutuskan untuk warna kulit tersangka dan membuka kasing. Saran untuk analisis asal genetika berhasil mencapai gelombang minat umum pada orang di masa lalu mereka sendiri. "Roots mania," yang disebut hobi ini dalam sebuah artikel di Time, dikhususkan untuk "obsesi terbaru Amerika" - penelitian genealogis.
Metode genom secara aktif digunakan oleh spesialis yang mempelajari asal usul dan evolusi manusia. Sebagai contoh, pada 2013, tim peneliti internasional menggunakan analisis genetik untuk membantah hipotesis tentang asal usul Yahudi Ashkenazi dari Khazar (
13 ). Kumpulan data genom yang digunakan oleh penulis berada dalam domain publik: berisi lebih dari 100 populasi dunia. Kami mengusulkan untuk mensimulasikan studi kecil dengan kami: untuk menentukan tempat pelanggan Genotek dalam sampel ini, dan pada saat yang sama untuk memahami rincian teknis penentuan populasi.
Tujuan penelitian
Identifikasi pelanggan Genotek di antara populasi referensi. Cari tahu apakah ada perwakilan Yahudi Ashkenazi dalam sampel kami. Tunjukkan prinsip dan metode analisis populasi seseorang.
Tujuan penelitian
Memproses data genotipe dari 722 subjek menggunakan program ADMIXTURE menggunakan set data dari Behar et al., 2013 sebagai sampel pelatihan.
Bahan dan Metode
Karya asli Behar et al., 2013, menggunakan data dari 1.774 orang: di antaranya adalah perwakilan dari 88 populasi non-Yahudi (dari Saudi, Asia Tengah, Asia Timur, Eropa, Timur Tengah, Afrika Utara, Siberia, Asia Selatan dan sub Afrika Sahara) dan 18 populasi Yahudi. Para penulis membutuhkan set data yang luas untuk secara akurat menentukan tempat ashkenaze dalam konteks populasi dunia: tugasnya adalah untuk menyajikan ketiga wilayah geografis di mana kelompok ini secara hipotesis dapat berasal dari - Eropa, Timur Tengah dan Khazar Khaganate. Para penulis menekankan perbedaan antara pendekatan pengambilan sampel, yang mewakili populasi Eropa modern, Timur Tengah dan Yahudi - keturunan langsung dari populasi leluhur, dan sampel yang sesuai dengan Khazar Kaganate, yang tidak ada lagi sekitar 1000 tahun yang lalu. Tangkapannya adalah bahwa tidak satu pun dari populasi yang ada adalah pewaris langsung ke Khaganate. Para penulis memilih penduduk Kaukasus Selatan (Abkhazia, Armenia, Azerbaijan, Georgia), Kaukasus Utara (Adygs, Balkars, Chechen, Kabardin, Ossetia dan beberapa negara lain), Chuvash, dan Tatar sebagai perwakilan modern dari Khazar.
Kami menambahkan sampel dari 722 orang dari berbagai wilayah Rusia ke set data.
Untuk analisis statistik, kami menggunakan program ADMIXTURE, yang memungkinkan kami memperkirakan asal individu yang paling mungkin berdasarkan data pada genotipe. Selain itu, penulis artikel yang dibahas menggunakan metode statistik lain yang memberikan jawaban yang mirip dengan pertanyaan yang diajukan. Kami akan fokus pada ADMIXTURE, karena algoritma inilah yang memungkinkan kami untuk memperkirakan kontribusi persentase populasi leluhur terhadap genom yang diteliti.
ADMIXTURE menggunakan metode Monte Carlo dalam rantai Markov (rantai Markov Monte Carlo, MCMC). Berikut ini adalah
tautan ke artikel oleh penulis algoritma untuk mereka yang ingin memahami secara lebih rinci sisi matematika dari proses tersebut.
Mari kita lihat bagaimana ADMIXTURE bekerja pada contoh sampel dan populasi dari set kami
Secara total, kami memiliki 2.496 sampel / individu, yang masing-masing milik salah satu dari 106 populasi modern. Kami menyarankan bahwa populasi modern kemungkinan besar adalah keturunan dari sejumlah kecil populasi leluhur. "Populasi leluhur" dalam analisis ini adalah beberapa kelompok genom kuno, disatukan oleh prinsip kesamaan genetik. ADMIXTURE memungkinkan keduanya untuk secara sewenang-wenang mengedepankan asumsi tentang jumlah cluster tersebut dalam sampel, dan untuk memilih angka optimal yang paling tepat menggambarkan distribusi nyata data genomik.
Setelah menerima informasi tentang genotipe dan perkiraan jumlah populasi "leluhur" (K), ADMIXTURE membangun model yang memperkirakan kontribusi masing-masing populasi "leluhur" untuk setiap sampel. Ketika menginterpretasikan data, komposisi kuantitatif genom (persentase kelompok) dan kualitatif adalah penting - ada atau tidaknya mereka dalam genom tertentu. Berdasarkan data ini, seseorang dapat membuat asumsi tentang proses evolusi dalam suatu populasi, khususnya, tentang ada atau tidak adanya "akar" umum dalam kelompok populasi. Namun, kesimpulan akan sah jika model yang kami buat baik: nilai optimal K. dipilih.
Kami memilih nilai optimal K
Bagaimana menentukan berapa banyak populasi "leluhur" yang paling cocok dengan yang sebenarnya untuk sampel yang diberikan? Secara empiris!
ADMIXTURE adalah program cerdas: membangun model struktur genetik populasi berdasarkan data pada genotipe individu (menilai kontribusi masing-masing kelompok genomik kuno untuk masing-masing genom sampel) untuk sejumlah K tertentu, dia tidak lupa membuat perbandingan dengan kenyataan di bagian akhir. Periksa seberapa baik input dijelaskan oleh model yang dibuat. Ukuran perbandingan adalah "kesalahan" - nilai yang menggambarkan ketidakcocokan antara model dan data nyata. Semakin besar kesalahan, semakin buruk asumsi jumlah populasi leluhur.
Bagaimana cara memilih nilai optimal K? Kami memulai algoritma ADMIXTURE pada sampel ini, menggantikan nilai-nilai K yang berbeda, dan kami memperoleh untuk masing-masing K nilai kesalahannya sendiri. Kami merencanakan ketergantungan besarnya kesalahan pada K. Berikut adalah grafik yang diperoleh oleh penulis artikel:
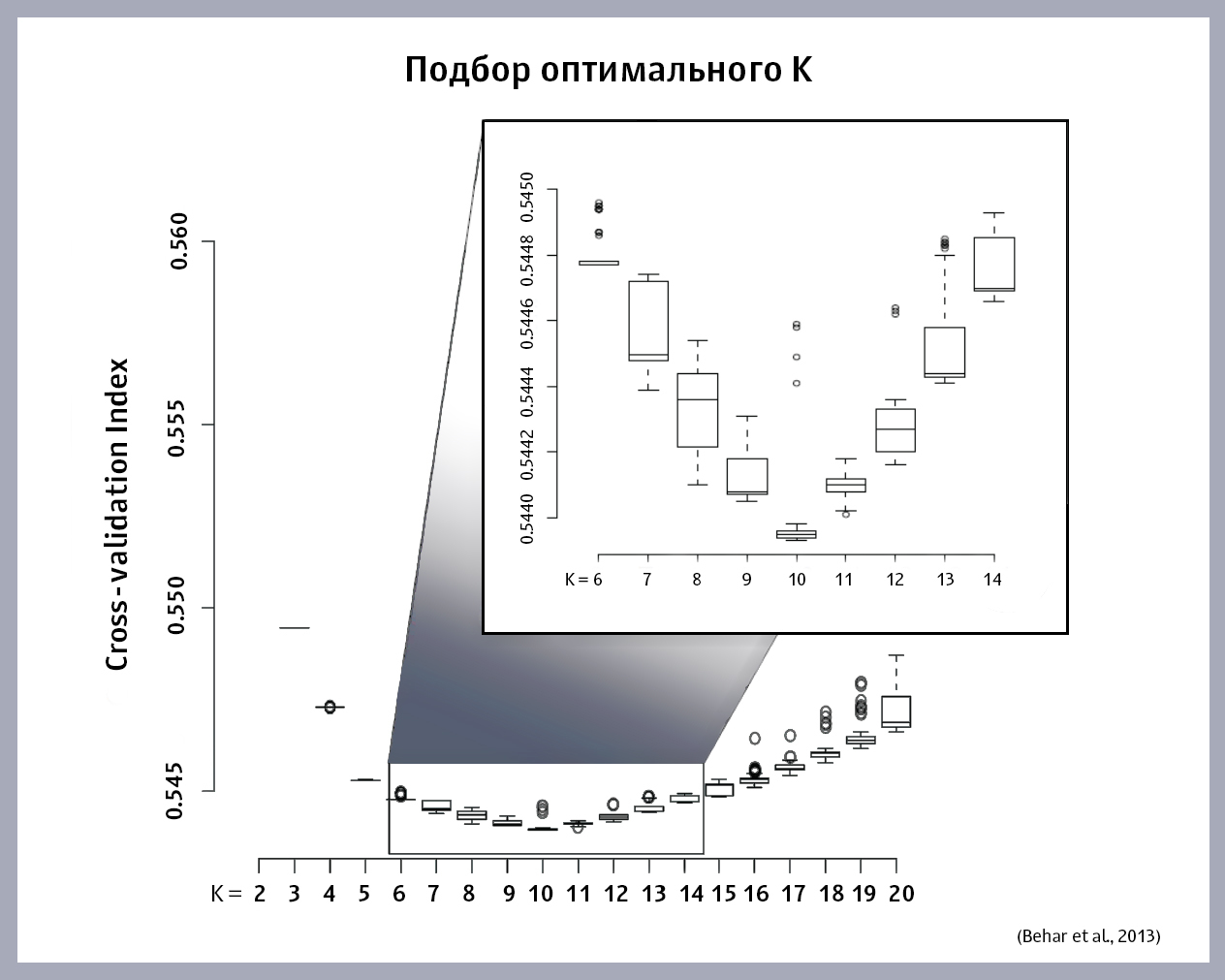
Nilai optimal K adalah pada titik minimum fungsi. Jika minimum tidak ditemukan pada grafik (fungsi terus-menerus bertambah atau berkurang), Anda harus membuat model dengan memilih Ks baru hingga Anda dapat menemukan yang tepat.
Bahkan dengan K yang dipilih secara optimal, keandalan hasil analisis tergantung pada kebenaran sampel:
1. Individu tidak boleh berhubungan satu sama lain.
2. Polimorfisme nukleotida tunggal (SNP) yang digunakan untuk genotipe harus didistribusikan secara merata di atas genom dengan kepadatan yang cukup tinggi.
3. Alel SNP harus berada dalam hubungan ekuilibrium, yaitu probabilitas keberadaan alel yang diberikan pada individu tertentu harus bergantung hanya pada frekuensi alel ini dalam populasi, dan bukan pada alel lain dalam genom.
Seperti yang dapat dilihat dari grafik, K optimal untuk sampel ini adalah 10 populasi "leluhur".
Hasil
Hasil analisis divisualisasikan oleh ADMIXTURE seperti ini (hanya sebagian data yang terlihat dalam gambar):
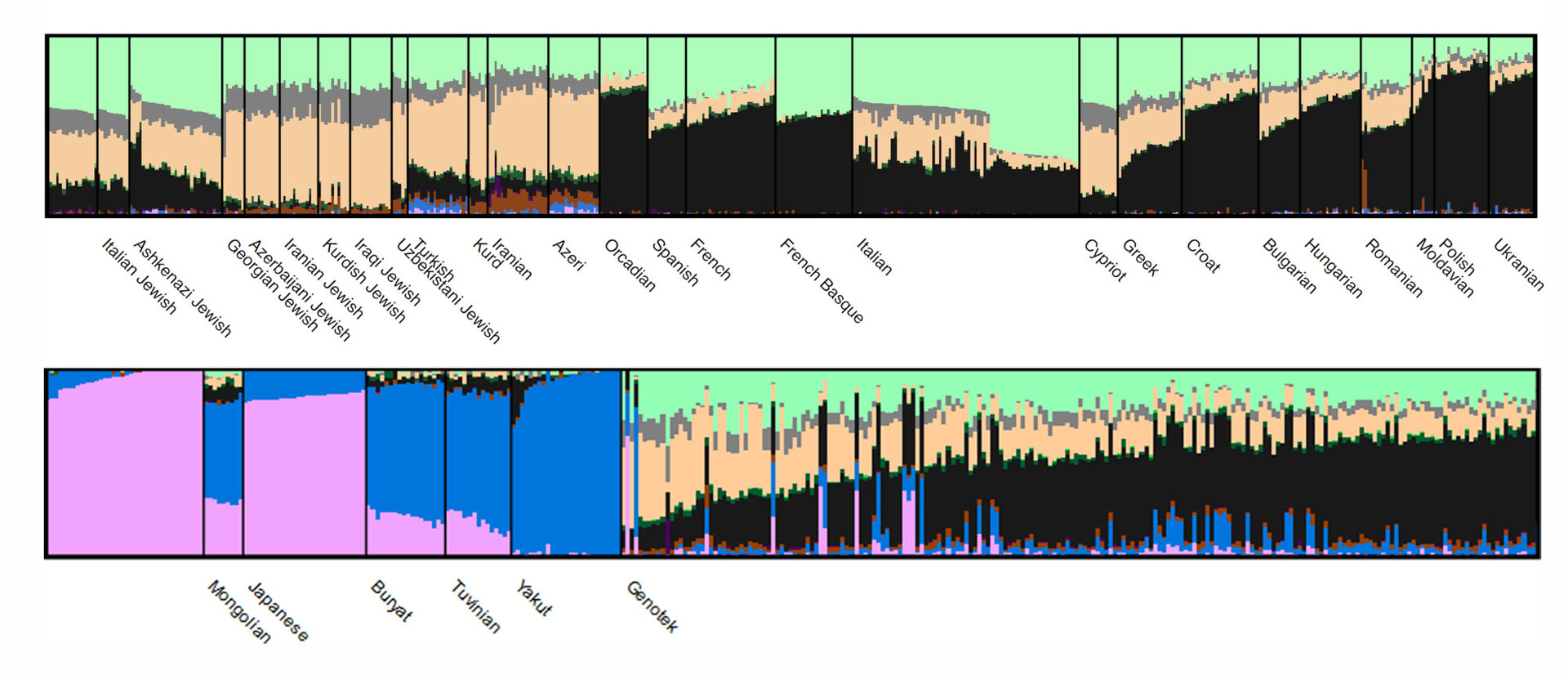
Setiap kelompok memiliki warna tersendiri, dan populasi berbeda (atau tidak berbeda) dalam bagian-bagian kelompok dalam genom.
Di sinilah letak versi interaktif gambar untuk studi terperinci: gerakkan mouse dan gulir untuk melihat semua populasi atau untuk mempertimbangkan beberapa kelompok secara lebih rinci.
Secara umum, dalam "populasi" Genotek, rasio klaster diharapkan sesuai dengan karakteristik pola populasi yang berasal dari Eropa Timur. Yang menarik dimulai pada tingkat sampel individu:
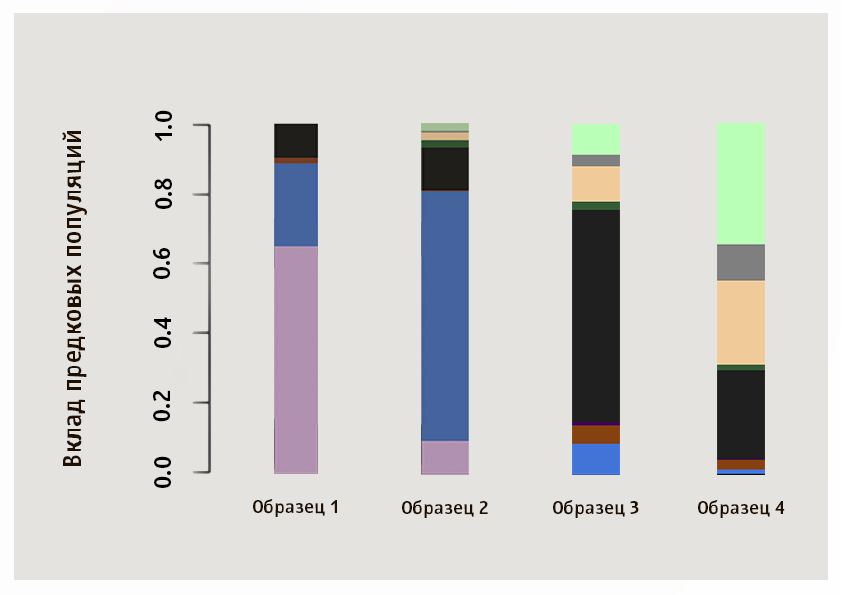
Meskipun populasi terdekat dengan sampel yang diberikan ditentukan oleh nilai numerik, banyak informasi juga dapat diperoleh dengan perbandingan pola secara visual. Kami menyarankan Anda menentukan sendiri populasi terdekat untuk sampel empat klien Genotek dari gambar.
JawabannyaDalam gambar ini, sampel 1 dan 2 berasal dari Asia: dominasi gugus merah muda khas untuk orang Jepang dan orang-orang Khan dalam sampel kami, biru untuk Yakuts, sampel ketiga menunjukkan rasio komponen khas Rusia, Belarusia, Ukraina dan Polandia, dan yang keempat khas Yahudi Ashkenaz.
Dari 722 sampel, kami menemukan 9 orang Yahudi Ashkenazi.
Kesimpulan
Afiliasi populasi jauh dari satu-satunya faktor yang menentukan identifikasi diri etnis seseorang. Namun, masih mungkin untuk mengungkapkan korelasi antara kelompok etnis dan struktur genom perwakilan mereka. Analisis semacam itu digunakan baik untuk tujuan ilmiah dan medis, dan untuk studi tentang akar mereka sendiri oleh semua pendatang. Pada saat yang sama, penting untuk memahami bahwa model terus ditingkatkan, dan hasil yang diperoleh untuk akurasi yang lebih besar harus dipertimbangkan dalam hubungannya dengan data lain, misalnya, pohon silsilah keluarga.
Tidak ada bukti yang ditemukan tentang asal-usul Khazar dari Ashkenazi oleh penulis artikel aslinya. Tes genetika, tentu saja, "tahu" bagaimana mengidentifikasi orang Yahudi - namun, orang tidak boleh lupa bahwa "Yahudi" adalah, pertama-tama, keadaan pikiran.
Dalam waktu dekat, tes DNA Silsilah yang diperbarui dengan hasil yang diperpanjang akan diluncurkan di Genotek: kami akan menambah jumlah populasi menjadi ratusan, tambah populasi Yahudi. Kami akan memperbarui informasi di akun pribadi Anda untuk semua orang yang pernah memberikan materi genetik kepada kami. Jika Anda masih belum genotip, kami mengundang Anda untuk
bergabung .
Referensi
- Foster M., Sharp R. (2002). Ras, Etnis, dan Genomik: Klasifikasi Sosial sebagai Proksi Heterogenitas Biologis. Genome Res.
- Collins FS, McKusick VA (2001). Implikasi dari Proyek Genom Manusia untuk ilmu kedokteran. JAMA.
- Nebert DW, Menon AG (2001) Farmakogenomik, etnis, dan gen kerentanan. Farmakogenomik J.
- Olden K., Guthrie J. (2001). Genomik: Implikasi untuk toksikologi. Mutat. Res.
- Yudell M., Roberts D., DeSalle R., Tishkoff S. (2016). Mengambil ras dari genetika manusia. Sains
- Ogura, Y. et al. (2001). Mutasi frameshift pada NOD2 terkait dengan kerentanan terhadap penyakit Crohn. Alam.
- Hugot, JP et al. (2001). Asosiasi varian berulang leusin NOD2 kaya dengan kerentanan terhadap penyakit Crohn. Alam.
- Inoue, N. (2002). Kurangnya varian NOD2 umum pada pasien Jepang dengan penyakit Crohn. Gastroenterologi.
- Martin, MP et al. (1998). Percepatan genetik perkembangan AIDS oleh varian promotor CCR5. Sains
- Gonzalez, E. et al. (1999). Efek pemodifikasi penyakit HIV-1 khusus ras terkait dengan haplotipe CCR5. Proc Natl Acad. Sci. USA
- Shi, Hong et al. (2009). Suhu Musim Dingin dan UV Sangat Terkait dengan Perubahan Genetik pada Jalur Penekan Tumor p53 di Asia Timur. American Journal of Human Genetics.
- Bamshad M., Wooding S., Salisbury B. et al. (2004). Mendekonstruksi hubungan antara genetika dan ras. Nat Rev Genet.
- Behar DM et al. (2013). Tidak Ada Bukti dari Data Genome-Wide dari Khazar Asal untuk Yahudi Ashkenazi. Biologi Manusia