Cara membaca artikel ini : Saya minta maaf atas kenyataan bahwa teksnya ternyata begitu panjang dan kacau. Untuk menghemat waktu Anda, saya memulai setiap bab dengan pengenalan "Apa yang Saya Pelajari," di mana saya menjelaskan esensi bab dalam satu atau dua kalimat.
"Tunjukkan solusinya!" Jika Anda hanya ingin melihat apa yang saya maksud, kemudian buka bab "Menjadi lebih inventif," tetapi saya pikir lebih menarik dan bermanfaat untuk membaca tentang kegagalan.Baru-baru ini, saya diperintahkan untuk mengatur proses untuk memproses sejumlah besar urutan DNA asli (secara teknis, ini adalah chip SNP). Itu perlu untuk mendapatkan data dengan cepat pada lokasi genetik yang diberikan (disebut SNP) untuk pemodelan berikutnya dan tugas-tugas lainnya. Dengan bantuan R dan AWK, saya dapat membersihkan dan mengatur data dengan cara alami, sangat mempercepat pemrosesan permintaan. Ini tidak mudah bagi saya dan membutuhkan banyak iterasi. Artikel ini akan membantu Anda menghindari beberapa kesalahan saya dan menunjukkan apa yang saya lakukan pada akhirnya.
Pertama, beberapa penjelasan pengantar.
Data
Pusat Pemrosesan Informasi Genetik Universitas kami telah memberi kami 25 TB data TSV. Saya membaginya menjadi 5 paket yang dikompres oleh Gzip, yang masing-masing berisi sekitar 240 file empat gigabyte. Setiap baris berisi data untuk satu SNP dari satu orang. Secara total, data ~ 2,5 juta SNP dan ~ 60 ribu orang dikirimkan. Selain informasi SNP, ada banyak kolom dalam file dengan angka yang mencerminkan berbagai karakteristik, seperti intensitas membaca, frekuensi alel yang berbeda, dll. Ada sekitar 30 kolom dengan nilai unik.
Tujuan
Seperti halnya proyek manajemen data, hal terpenting adalah menentukan bagaimana data akan digunakan. Dalam hal ini,
sebagian besar, kami akan memilih model dan alur kerja untuk SNP berdasarkan SNP . Artinya, pada saat yang sama kita akan membutuhkan data hanya untuk satu SNP. Saya harus belajar cara mengekstrak semua catatan yang terkait dengan salah satu dari 2,5 juta SNP sesederhana mungkin, lebih cepat dan lebih murah.
Bagaimana tidak melakukannya
Saya akan mengutip klise yang sesuai:
Saya tidak gagal ribuan kali, saya baru saja menemukan seribu cara untuk tidak menguraikan banyak data dalam format yang sesuai untuk permintaan.
Upaya pertama
Apa yang saya pelajari : Tidak ada cara murah untuk mengurai 25 TB sekaligus.
Setelah mendengarkan subjek "Metode Pengolahan Data Besar Lanjut" di Vanderbilt University, saya yakin itu adalah topi. Mungkin akan membutuhkan satu atau dua jam untuk mengkonfigurasi server Hive untuk menjalankan semua data dan melaporkan hasilnya. Karena data kami disimpan di AWS S3, saya menggunakan layanan
Athena , yang memungkinkan Anda untuk menerapkan kueri Hive SQL ke data S3. Tidak perlu mengkonfigurasi / menaikkan Hive-cluster, dan bahkan hanya membayar untuk data yang Anda cari.
Setelah saya menunjukkan data dan formatnya kepada Athena, saya melakukan beberapa tes dengan pertanyaan serupa:
select * from intensityData limit 10;
Dan dengan cepat mendapat hasil yang terstruktur dengan baik. Selesai
Sampai kami mencoba menggunakan data dalam pekerjaan ...
Saya diminta untuk mengeluarkan semua informasi SNP untuk menguji model di atasnya. Saya menjalankan kueri:
select * from intensityData where snp = 'rs123456';
... dan menunggu. Setelah delapan menit dan lebih dari 4 TB data yang diminta, saya mendapatkan hasilnya. Athena membebankan biaya untuk jumlah data yang ditemukan, sebesar $ 5 per terabyte. Jadi permintaan tunggal ini biaya $ 20 dan delapan menit menunggu. Untuk menjalankan model sesuai dengan semua data, perlu menunggu 38 tahun dan membayar $ 50 juta. Jelas, ini tidak cocok untuk kita.
Itu perlu untuk menggunakan Parket ...
Apa yang saya pelajari : Hati-hati dengan ukuran file Parket Anda dan organisasi mereka.
Pada awalnya saya mencoba untuk memperbaiki situasi dengan mengubah semua TSV ke
file Parket . Mereka nyaman untuk bekerja dengan set data besar, karena informasi di dalamnya disimpan dalam bentuk kolom: setiap kolom terletak di segmen memori / disk sendiri, tidak seperti file teks di mana baris berisi elemen dari setiap kolom. Dan jika Anda perlu menemukan sesuatu, maka baca saja kolom yang diperlukan. Selain itu, rentang nilai disimpan di setiap file dalam kolom, jadi jika nilai yang diinginkan tidak berada dalam rentang kolom, Spark tidak akan membuang waktu memindai seluruh file.
Saya menjalankan tugas
Lem AWS sederhana untuk mengkonversi TSV kami ke Parket dan menjatuhkan file baru ke Athena. Butuh sekitar 5 jam. Tetapi ketika saya meluncurkan permintaan, dibutuhkan waktu yang hampir bersamaan dan sedikit uang untuk menyelesaikannya. Faktanya adalah Spark, yang mencoba mengoptimalkan tugas, cukup membongkar satu TSV-chunk dan meletakkannya di Parquet-chunk sendiri. Dan karena setiap potongan cukup besar dan berisi catatan lengkap banyak orang, semua SNP disimpan dalam setiap file, sehingga Spark harus membuka semua file untuk mengekstrak informasi yang diperlukan.
Anehnya, tipe kompresi default (dan disarankan) di Parket - tajam - tidak dapat dipisahkan. Oleh karena itu, setiap pelaksana terjebak pada tugas membongkar dan mengunduh dataset 3,5 GB penuh.

Kami mengerti masalahnya
Apa yang saya pelajari : menyortir itu sulit, terutama jika datanya didistribusikan.
Tampak bagi saya bahwa sekarang saya mengerti esensi masalah. Yang harus saya lakukan adalah mengurutkan data berdasarkan kolom SNP, bukan oleh orang. Kemudian beberapa SNP akan disimpan dalam potongan data yang terpisah, dan kemudian fungsi pintar Parket "terbuka hanya jika nilainya dalam kisaran" akan memanifestasikan dirinya dalam semua kemuliaan. Sayangnya, memilah miliaran baris yang tersebar di sebuah cluster telah terbukti menjadi tugas yang menakutkan.
AWS tentu saja tidak ingin mengembalikan uang itu karena "Saya seorang siswa yang linglung." Setelah saya mulai menyortir di Lem Amazon, itu bekerja selama 2 hari dan jatuh.
Bagaimana dengan mempartisi?
Apa yang saya pelajari : Partisi dalam Spark harus seimbang.
Kemudian muncul ide untuk membagi data pada kromosom. Ada 23 di antaranya (dan beberapa lagi, diberi DNA mitokondria dan area yang belum dipetakan).
Ini akan memungkinkan Anda untuk membagi data menjadi bagian-bagian yang lebih kecil. Jika Anda hanya menambahkan satu baris
partition_by = "chr" ke fungsi ekspor Spark di skrip Glue, maka data harus diurutkan menjadi kotak.
 Genom terdiri dari banyak fragmen yang disebut kromosom.
Genom terdiri dari banyak fragmen yang disebut kromosom.Sayangnya, ini tidak berhasil. Kromosom memiliki ukuran yang berbeda, dan karena itu jumlah informasi yang berbeda. Ini berarti bahwa tugas-tugas yang dikirim Spark ke pekerja tidak seimbang dan dilakukan perlahan-lahan, karena beberapa node selesai sebelumnya dan menganggur. Namun, tugas itu selesai. Tetapi ketika meminta satu SNP, ketidakseimbangan kembali menyebabkan masalah. Biaya pemrosesan SNP pada kromosom yang lebih besar (yaitu, tempat kami ingin mendapatkan data) menurun hanya sekitar 10 kali. Banyak, tetapi tidak cukup.
Dan jika Anda membaginya menjadi partisi yang lebih kecil?
Apa yang saya pelajari : tidak pernah mencoba melakukan 2,5 juta partisi sama sekali.
Saya memutuskan untuk berjalan-jalan dan mempartisi setiap SNP. Ini menjamin ukuran partisi yang sama.
BURUK IDE . Saya mengambil keuntungan dari Lem dan menambahkan
partition_by = 'snp' tidak bersalah. Tugas dimulai dan mulai dijalankan. Sehari kemudian, saya memeriksa dan melihat bahwa tidak ada yang ditulis dalam S3 sejauh ini, jadi saya membunuh tugas itu. Sepertinya Lem sedang menulis file perantara ke tempat tersembunyi di S3, dan banyak file, mungkin beberapa juta. Akibatnya, kesalahan saya menelan biaya lebih dari seribu dolar dan tidak menyenangkan mentor saya.
Partisi + penyortiran
Apa yang saya pelajari : menyortir masih sulit, seperti menyiapkan Spark.
Upaya terakhir untuk mempartisi adalah saya mempartisi kromosom dan kemudian mengurutkan setiap partisi. Secara teori, ini akan mempercepat setiap permintaan, karena data SNP yang diinginkan harus dalam beberapa potongan Parket dalam kisaran yang diberikan. Sayangnya, menyortir bahkan data yang dipartisi telah terbukti menjadi tugas yang sulit. Akibatnya, saya beralih ke ESDM untuk kluster khusus dan menggunakan delapan instance kuat (C5.4xl) dan Sparklyr untuk membuat alur kerja yang lebih fleksibel ...
# Sparklyr snippet to partition by chr and sort w/in partition # Join the raw data with the snp bins raw_data group_by(chr) %>% arrange(Position) %>% Spark_write_Parquet( path = DUMP_LOC, mode = 'overwrite', partition_by = c('chr') )
... namun, tugas itu masih belum selesai. Saya menyetel dengan segala cara: Saya meningkatkan alokasi memori untuk setiap pelaksana kueri, menggunakan node dengan jumlah memori yang besar, menggunakan variabel penyiaran, tetapi setiap kali ternyata menjadi setengah ukuran, dan lambat laun para pemain mulai gagal, sampai semuanya berhenti.
Saya semakin inventif
Apa yang saya pelajari : terkadang data khusus memerlukan solusi khusus.
Setiap SNP memiliki nilai posisi. Ini adalah angka yang sesuai dengan jumlah basa yang terletak di sepanjang kromosomnya. Ini adalah cara yang baik dan alami untuk mengatur data kami. Pada awalnya saya ingin mempartisi berdasarkan wilayah setiap kromosom. Misalnya, posisi 1 - 2000, 2001 - 4000, dll. Tetapi masalahnya adalah SNP tidak terdistribusi secara merata di seluruh kromosom, oleh karena itu ukuran kelompok akan sangat bervariasi.
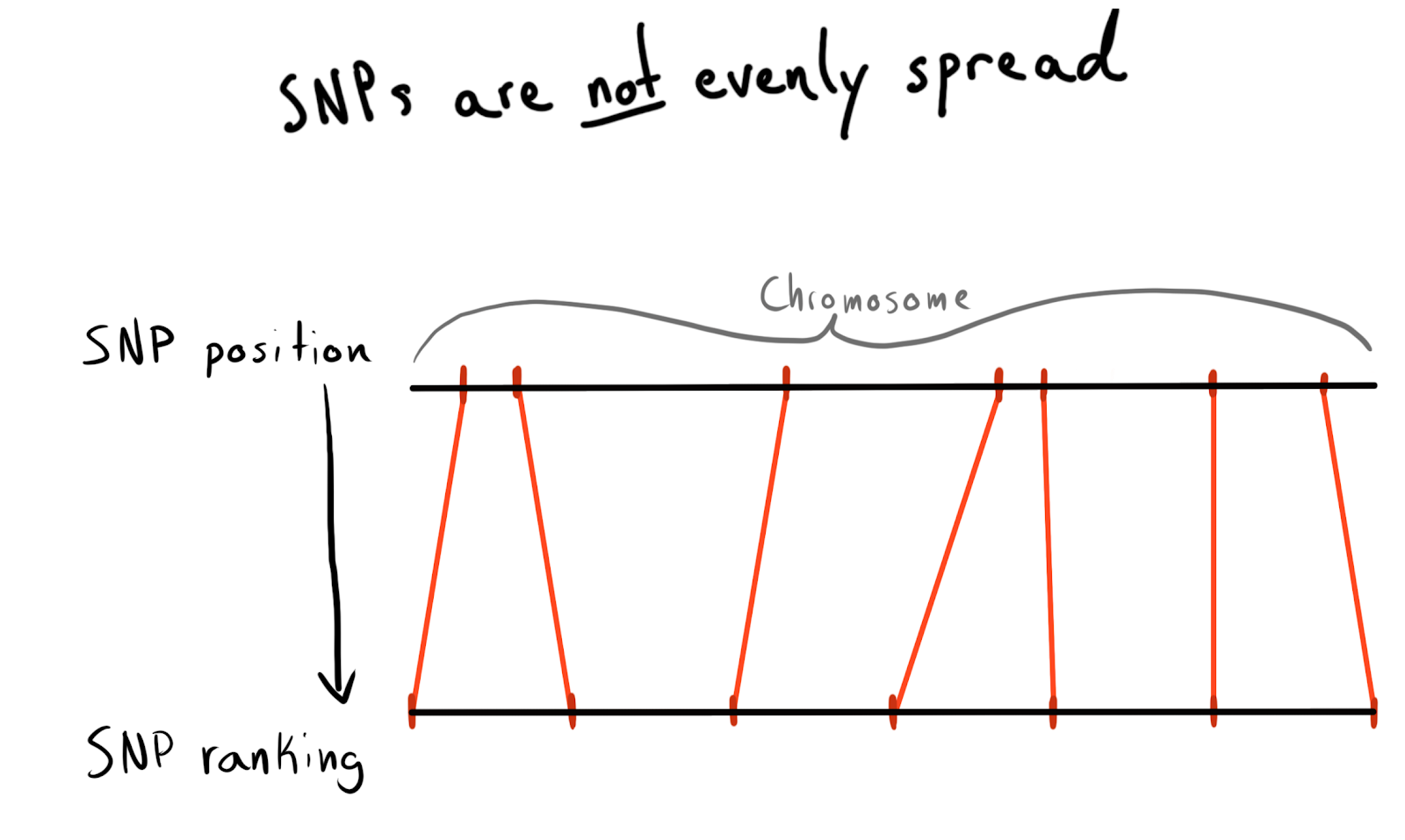
Sebagai hasilnya, saya kemudian dibagi menjadi beberapa kategori (peringkat) posisi. Menurut data yang sudah diunduh, saya menjalankan permintaan daftar SNP unik, posisi dan kromosom mereka. Kemudian dia mengurutkan data di dalam setiap kromosom dan mengumpulkan SNP menjadi kelompok (bin) dengan ukuran tertentu. Katakan masing-masing 1.000 SNP. Ini memberi saya hubungan SNP dengan kelompok-dalam-kromosom.
Pada akhirnya, saya membuat grup (bin) pada 75 SNP, saya akan menjelaskan alasannya di bawah ini.
snp_to_bin <- unique_snps %>% group_by(chr) %>% arrange(position) %>% mutate( rank = 1:n() bin = floor(rank/snps_per_bin) ) %>% ungroup()
Pertama coba dengan Spark
Apa yang saya pelajari : Integrasi spark cepat, tetapi mempartisi masih mahal.
Saya ingin membaca bingkai data kecil (2,5 juta baris) ini di Spark, menggabungkannya dengan data mentah, dan kemudian mempartisi dengan kolom
bin baru ditambahkan.
Saya menggunakan
sdf_broadcast() , jadi Spark mengetahui bahwa itu harus mengirim bingkai data ke semua node. Ini berguna jika datanya kecil dan diperlukan untuk semua tugas. Jika tidak, Spark mencoba menjadi pintar dan mendistribusikan data sesuai kebutuhan, yang dapat menyebabkan rem.
Dan lagi, ide saya tidak berhasil: tugas-tugasnya bekerja sebentar, menyelesaikan merger, dan kemudian, seperti para pelaksana yang diluncurkan dengan mempartisi, mereka mulai gagal.
Tambahkan AWK
Apa yang saya pelajari : jangan tidur ketika dasar mengajarkan Anda. Tentunya seseorang sudah memecahkan masalah Anda di tahun 1980-an.
Hingga saat ini, penyebab semua kegagalan saya dengan Spark adalah kebingungan data di cluster. Mungkin situasinya dapat ditingkatkan dengan pra-pemrosesan. Saya memutuskan untuk mencoba membagi data teks mentah menjadi kolom kromosom, jadi saya berharap untuk memberikan Spark dengan data "pra-partisi".
Saya mencari di StackOverflow untuk cara memecah nilai kolom dan menemukan
jawaban yang bagus. Menggunakan AWK, Anda dapat membagi file teks menjadi nilai kolom dengan menulis ke skrip, daripada mengirim hasilnya ke
stdout .
Untuk pengujian, saya menulis skrip Bash. Saya mengunduh salah satu TSV yang dikemas, kemudian membukanya dengan
gzip dan mengirimkannya ke
awk .
gzip -dc path/to/chunk/file.gz | awk -F '\t' \ '{print $1",..."$30">"chunked/"$chr"_chr"$15".csv"}'
Berhasil!
Mengisi inti
Apa yang saya pelajari :
gnu parallel adalah hal yang ajaib, semua orang harus menggunakannya.
Pemisahan itu agak lambat, dan ketika saya mulai
htop untuk menguji penggunaan contoh EC2 yang kuat (dan mahal), ternyata saya hanya menggunakan satu inti dan sekitar 200 MB memori. Untuk menyelesaikan masalah dan tidak kehilangan banyak uang, perlu dipikirkan cara memparalelkan pekerjaan. Untungnya, dalam
Data Science Jeron Janssens yang menakjubkan
di buku Command Line , saya menemukan bab tentang paralelisasi. Dari sini saya belajar tentang
gnu parallel , metode yang sangat fleksibel untuk mengimplementasikan multithreading di Unix.
Ketika saya memulai partisi menggunakan proses baru, semuanya baik-baik saja, tetapi ada hambatan - mengunduh objek S3 ke disk tidak terlalu cepat dan tidak sepenuhnya paralel. Untuk memperbaikinya, saya melakukan ini:
- Saya menemukan bahwa adalah mungkin untuk mengimplementasikan langkah S3-unduh langsung di dalam pipa, sepenuhnya menghilangkan penyimpanan perantara pada disk. Ini berarti bahwa saya dapat menghindari penulisan data mentah ke disk dan menggunakan lebih kecil, dan karenanya penyimpanan lebih murah di AWS.
- Perintah
aws configure set default.s3.max_concurrent_requests 50 sangat meningkatkan jumlah utas yang digunakan AWS CLI (ada 10 secara default).
- Saya beralih ke instance EC2 yang dioptimalkan untuk kecepatan jaringan, dengan huruf n pada namanya. Saya menemukan bahwa hilangnya daya komputasi saat menggunakan n-instances lebih dari diimbangi dengan peningkatan kecepatan pengunduhan. Untuk sebagian besar tugas, saya menggunakan c5n.4xl.
- Saya mengubah
gzip menjadi pigz , ini adalah alat gzip yang dapat melakukan hal-hal keren untuk memparalelkan tugas membongkar file yang awalnya tidak tertandingi (ini paling sedikit membantu).
Langkah-langkah ini dikombinasikan satu sama lain sehingga semuanya bekerja sangat cepat. Berkat peningkatan kecepatan pengunduhan dan penolakan penulisan ke disk, sekarang saya dapat memproses paket 5-terabyte hanya dalam beberapa jam.
Tweet ini seharusnya menyebutkan 'TSV'. Sayang
Menggunakan data yang diurai ulang
Apa yang saya pelajari : Spark menyukai data yang tidak terkompresi dan tidak suka menggabungkan partisi.
Sekarang datanya dalam S3 dalam format yang sudah dibongkar (baca, bagikan) dan semi-teratur, dan saya bisa kembali ke Spark lagi. Kejutan menunggu saya: Saya kembali gagal mencapai yang diinginkan! Sangat sulit untuk memberi tahu Spark bagaimana tepatnya data dipartisi. Dan bahkan ketika saya melakukan ini, ternyata ada terlalu banyak partisi (95 ribu), dan ketika saya mengurangi jumlah mereka menjadi batas yang koheren dengan
coalesce , itu merusak partisi saya. Saya yakin ini bisa diperbaiki, tetapi dalam beberapa hari pencarian, saya tidak dapat menemukan solusi. Pada akhirnya, saya menyelesaikan semua tugas di Spark, meskipun butuh beberapa saat, dan file parket saya tidak terlalu kecil (~ 200 Kb). Namun, data di tempat itu dibutuhkan.
 Terlalu kecil dan berbeda, luar biasa!
Terlalu kecil dan berbeda, luar biasa!Menguji permintaan Spark lokal
Apa yang saya pelajari : Spark memiliki terlalu banyak masalah dalam menyelesaikan masalah sederhana.
Dengan mengunduh data dalam format cerdas, saya dapat menguji kecepatannya. Saya mengatur skrip pada R untuk memulai server Spark lokal, dan kemudian saya memuat frame data Spark dari repositori tertentu dari grup Parket (bin). Saya mencoba memuat semua data, tetapi tidak bisa membuat Sparklyr mengenali partisi.
sc <- Spark_connect(master = "local") desired_snp <- 'rs34771739' # Start a timer start_time <- Sys.time() # Load the desired bin into Spark intensity_data <- sc %>% Spark_read_Parquet( name = 'intensity_data', path = get_snp_location(desired_snp), memory = FALSE ) # Subset bin to snp and then collect to local test_subset <- intensity_data %>% filter(SNP_Name == desired_snp) %>% collect() print(Sys.time() - start_time)
Eksekusi memakan waktu 29,415 detik. Jauh lebih baik, tetapi tidak terlalu baik untuk pengujian massal apa pun. Selain itu, saya tidak bisa mempercepat pekerjaan menggunakan caching, karena ketika saya mencoba untuk men-cache bingkai data dalam memori, Spark selalu macet, bahkan ketika saya mengalokasikan lebih dari 50 GB memori untuk dataset yang beratnya kurang dari 15.
Kembali ke AWK
Apa yang saya pelajari : array asosiatif AWK sangat efisien.
Saya mengerti bahwa saya bisa mencapai kecepatan yang lebih tinggi. Saya ingat bahwa dalam panduan AWK
Bruce Barnett yang sangat baik, saya membaca tentang fitur keren yang disebut “
array asosiatif ”. Sebenarnya, ini adalah pasangan kunci-nilai, yang karena alasan tertentu disebut berbeda di AWK, dan karena itu saya entah bagaimana tidak secara khusus menyebutkannya.
Roman Cheplyaka mengingat bahwa istilah “array asosiatif” jauh lebih tua daripada istilah “pasangan nilai kunci”. Bahkan jika Anda
mencari nilai kunci di Google Ngram , Anda tidak akan melihat istilah ini di sana, tetapi Anda akan menemukan array asosiatif! Selain itu, pasangan kunci-nilai paling sering dikaitkan dengan basis data, sehingga jauh lebih logis untuk membandingkan dengan hashmap. Saya menyadari bahwa saya bisa menggunakan array asosiatif ini untuk menghubungkan SNP saya ke tabel bin dan data mentah tanpa menggunakan Spark.
Untuk ini, dalam skrip AWK, saya menggunakan blok
BEGIN . Ini adalah bagian dari kode yang dieksekusi sebelum baris pertama data ditransfer ke badan utama skrip.
join_data.awk BEGIN { FS=","; batch_num=substr(chunk,7,1); chunk_id=substr(chunk,15,2); while(getline < "snp_to_bin.csv") {bin[$1] = $2} } { print $0 > "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv" }
Perintah
while(getline...) memuat semua baris dari grup CSV (nampan), mengatur kolom pertama (nama SNP) sebagai kunci untuk array asosiatif
bin dan nilai kedua (grup) sebagai nilainya. Kemudian, di blok
{ } , yang diterapkan ke semua baris file utama, setiap baris dikirim ke file output, yang mendapatkan nama unik tergantung pada grupnya (bin):
..._bin_"bin[$1]"_...batch_num dan
chunk_id sesuai dengan data yang disediakan oleh pipeline, yang menghindari status ras, dan setiap utas eksekusi yang diluncurkan secara
parallel menulis ke file uniknya sendiri.
Karena saya menyebarkan semua data mentah ke folder pada kromosom yang tersisa setelah percobaan saya sebelumnya dengan AWK, sekarang saya bisa menulis skrip Bash lain untuk diproses pada kromosom sekaligus dan memberikan data yang dipartisi lebih dalam ke S3.
DESIRED_CHR='13'
Script memiliki dua bagian
parallel .
Bagian pertama membaca data dari semua file yang berisi informasi tentang kromosom yang diinginkan, kemudian data ini didistribusikan di seluruh aliran yang menyebarkan file ke grup yang sesuai (bin). Untuk mencegah kondisi balapan terjadi ketika beberapa aliran ditulis ke satu file, AWK mentransfer nama file untuk menulis data ke tempat yang berbeda, misalnya,
chr_10_bin_52_batch_2_aa.csv . Akibatnya, banyak file kecil dibuat pada disk (untuk ini saya menggunakan volume EBS terabyte).
Pipa dari bagian
parallel kedua melewati kelompok (bin) dan menggabungkan file masing-masing ke CSV umum dengan
cat , dan kemudian mengirimkannya untuk diekspor.
Disiarkan ke R?
Apa yang saya pelajari : Anda dapat mengakses
stdin dan
stdout dari skrip R, dan karenanya menggunakannya dalam pipeline.
Dalam skrip Bash, Anda mungkin memperhatikan baris ini:
...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... Ini menerjemahkan semua file grup gabungan (nampan) ke dalam skrip R di bawah ini.
{} adalah teknik
parallel khusus yang memasukkan data apa pun yang dikirim olehnya ke aliran yang ditentukan langsung ke perintah itu sendiri. Opsi
{#} memberikan ID utas unik, dan
{%} mewakili nomor slot pekerjaan (diulangi, tetapi tidak pernah secara bersamaan). Daftar semua opsi dapat ditemukan di
dokumentasi. #!/usr/bin/env Rscript library(readr) library(aws.s3) # Read first command line argument data_destination <- commandArgs(trailingOnly = TRUE)[1] data_cols <- list(SNP_Name = 'c', ...) s3saveRDS( read_csv( file("stdin"), col_names = names(data_cols), col_types = data_cols ), object = data_destination )
Ketika variabel
file("stdin") diteruskan ke
readr::read_csv , data yang diterjemahkan ke dalam skrip-R dimuat ke dalam bingkai, yang kemudian ditulis langsung ke S3 sebagai file
aws.s3 menggunakan
aws.s3 .
RDS sedikit seperti versi Parquet yang lebih muda, tanpa embel-embel penyimpanan kolom.
Setelah menyelesaikan skrip Bash, saya menerima
.rds file
.rds terletak pada S3, yang memungkinkan saya untuk menggunakan kompresi yang efisien dan tipe
.rds .
Meski menggunakan rem R, semuanya bekerja sangat cepat. Tidak mengherankan bahwa fragmen pada R yang bertanggung jawab untuk membaca dan menulis data dioptimalkan dengan baik. Setelah menguji pada satu kromosom berukuran sedang, tugas diselesaikan pada contoh C5n.4xl dalam waktu sekitar dua jam.
Batasan S3
Apa yang saya pelajari : berkat implementasi jalur yang cerdas, S3 dapat memproses banyak file.
Saya khawatir jika S3 bisa menangani banyak file yang ditransfer ke sana. Saya bisa membuat nama file bermakna, tetapi bagaimana S3 akan mencari mereka?
 S3 ,
S3 , / . FAQ- S3., S3 - . (bucket) , — .
Amazon, , «-----» . : get-, . , 20 . bin-. , , (, , ). .
?
: — .
: « ?» ( gzip CSV- 7 ) . , R Parquet ( Arrow) Spark. R, , , .
: , .
, .
EC2 , ( , Spark ). , , AWS- 10 .
R .
S3 , .
library(aws.s3) library(tidyverse) chr_sizes <- get_bucket_df( bucket = '...', prefix = '...', max = Inf ) %>% mutate(Size = as.numeric(Size)) %>% filter(Size != 0) %>% mutate(
, , ,
num_jobs , .
num_jobs <- 7
purrr .
1:1000 %>% map_df(shuffle_job) %>% filter(sd == min(sd)) %>% pull(data) %>% pluck(1)
, . Bash-
for . 10 . , . , .
for DESIRED_CHR in "16" "9" "7" "21" "MT" do
:
sudo shutdown -h now
… ! AWS CLI
user_data Bash- . , .
aws ec2 run-instances ...\ --tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]" \ --user-data file://<<job_script_loc>>
!
: API .
- . , . API .
.rds Parquet-, , . R-.
, ,
get_snp .
pkgdown , .
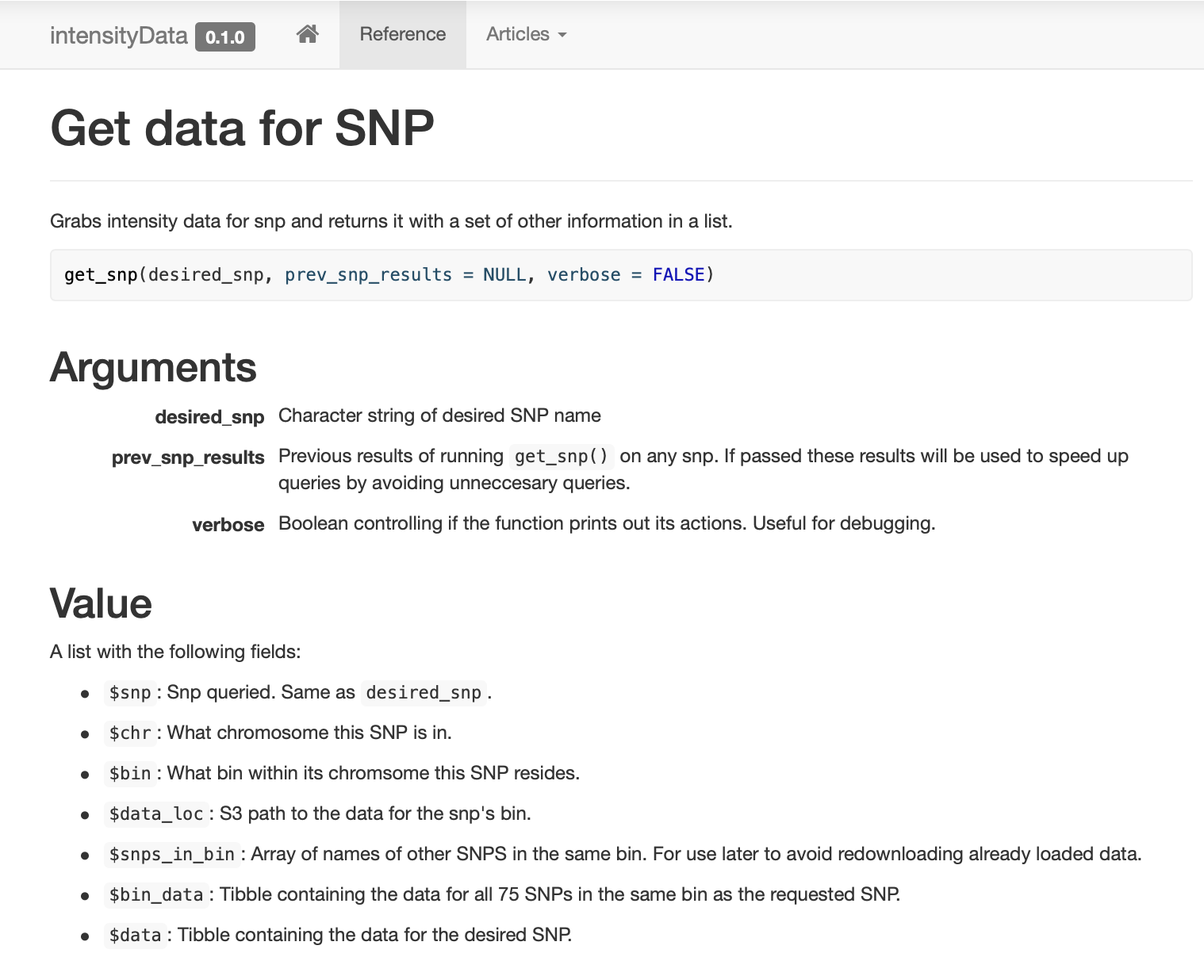
: , !
SNP , (binning) . SNP, (bin). ( ) .
, . , . ,
dplyr::filter , , .
,
prev_snp_results snps_in_bin . SNP (bin), , . SNP (bin) :
Hasil
( ) , . , . .
, , , …
. . ( ), , (bin) , SNP 0,1 , , S3 .
Kesimpulan
— . , . , . , , , . , , , , . , , , , - .
. , , «» , . .
:
- 25 ;
- Parquet- ;
- Spark ;
- 2,5 ;
- , Spark;
- ;
- Spark , ;
- , , - 1980-;
gnu parallel — , ;
- Spark ;
- Spark ;
- AWK ;
stdin stdout R-, ;
- S3 ;
- — ;
- , ;
- API ;
- , !