Dalam
artikel terperinci sebelumnya tentang
Genom Lengkap, kami berjanji untuk menerbitkan tiga masalah dan memberikan tes kepada orang yang pertama-tama memecahkan ketiganya dengan benar. Pada saat yang sama, kami memberikan contoh bagaimana bekerja dengan data genetik dalam tugas-tugas ini. Hari ini kami menerbitkan yang pertama.

Pada
artikel pertama
, kami berbagi informasi dan tautan bermanfaat yang berguna untuk bekerja dengan data bioinformatika. Kami sarankan Anda membacanya terlebih dahulu jika Anda melewatkannya.
PenafianBekerja dengan data genetik dilakukan pada sistem Unix (Linux, macOS), karena beberapa perintah dan perangkat lunak tidak tersedia di Windows. Oleh karena itu, untuk pengguna Windows, salah satu solusi paling sederhana adalah menyewa mesin virtual Linux.
Semua operasi yang dijelaskan di bawah ini dilakukan pada baris perintah - terminal. Sebelum Anda mulai, pelajari cara bekerja di terminal yang menjalankan OS Anda dan gunakan perintah, karena beberapa di antaranya berpotensi membahayakan OS dan data Anda.
Perangkat Lunak yang Diperlukan
Kami telah mengumpulkan
gambar mesin virtual (VM) dengan semua perangkat lunak yang diperlukan di Yandex.Cloud. Daftarkan di Yandex.Cloud, di akun Anda di bagian Compute Cloud, klik Buat VM. Sebagai gambar publik, pilih 1000 Genom dari katalog Analisis Data Atlas.
Konfigurasi VM: 100% 2vCPU, 8GB RAM, 20GB HDD. Saat membuat VM, Anda harus memasukkan data pembayaran, tetapi tidak ada yang dihapuskan dari akun. Awal dan hibah tambahan untuk kata kode sudah cukup untuk bekerja dengan VM dan gambar dari Atlas hingga 31 Desember 2019 secara gratis. Untuk menerima dana untuk menyelesaikan tugas, kirim kata kode "ATLAS" ke
dukungan Yandex.Cloud .
Catatan: hibah ini berlaku untuk pengguna Yandex.Cloud baru yang telah mendaftar sejak 18 Desember 2019 atau bagi mereka yang masih memiliki masa uji coba dan memiliki hibah awal. Kata kode ATLAS hanya valid satu kali.
Pertama buat kunci ssh di komputer lokal tempat Anda berencana untuk terhubung ke VM:
ssh-keygen -o -t rsa -b 4096 -C "my-local-machine" -f ~/.ssh/yandex-cloud -a 100
Jangan lupa menyalin isi file
~/.ssh/yandex-cloud.pub ke jendela yang sesuai saat membuat VM.
Jika Anda ingin menginstal perangkat lunak di komputer Anda, di bawah ini adalah semua informasi instalasi. Jika Anda memutuskan untuk menggunakan Yandex.Cloud, buat VM dan lanjutkan ke bagian berikutnya.
Plink
Plink adalah paket perangkat lunak untuk memanipulasi data genetik dan pencarian asosiasi genome luas (GWAS). Ini dikembangkan oleh ahli genetika Sean Purcell (Shaun Purcell). Sejak 2008, dengan bantuan Plink, ratusan GWAS telah dilakukan di seluruh dunia, hasil yang terbaik yang digunakan Atlas sebagai sumber data untuk algoritme untuk menghitung risiko penyakit.
Plink menawarkan seperangkat alat untuk menyimpan dan mengonversi data genotip dan mencarinya. Plink juga memungkinkan untuk pemrosesan statistik, analisis linkage disequilibrium (LD), identitas dengan keturunan (IBD) dan analisis identitas (status oleh IBS), stratifikasi populasi dan tes epistasis - interaksi beberapa variasi genetik di antara mereka sendiri.
IBD dan IBS digunakan untuk menganalisis komposisi populasi dan menentukan hubungan kekerabatan.
Contoh epistasis adalah variasi rs7412 dan rs429358 pada gen APOE, kombinasi varian tertentu yang secara tajam meningkatkan risiko pengembangan penyakit Alzheimer, sementara masing-masing varian secara individual hanya memberikan kontribusi kecil terhadap risiko.
Unduh Plink versi stabil dari situs
web resmi.
BCFtools
BCFtools adalah seperangkat utilitas untuk memanipulasi data genetik dalam format VCF dan mitra binernya BCF. Daftar kemungkinan aplikasi paket BCFtools termasuk anotasi, pemfilteran, penggabungan dan pemisahan file VCF / BCF, menemukan persimpangan, pengindeksan, pencarian selektif, pengurutan, penghitungan statistik, dll.
Untuk menginstal, lakukan:
git clone git://github.com/samtools/htslib.git git clone git://github.com/samtools/bcftools.git cd bcftools
Proses instalasi dijelaskan secara lebih rinci di
sini .
RAJA
Paket KING (Inferensi berbasis kekerabatan untuk Gwas) digunakan dalam studi populasi ketika bekerja dengan data dari pencarian genome untuk asosiasi untuk menentukan hubungan kekerabatan dalam data yang dipelajari. Dalam tugas ini, RA akan membantu menentukan tingkat kekerabatan beberapa sampel dari proyek 1000 Genom.
Anda dapat mengunduhnya di
sini . Untuk mengatasi masalah, manual RA tersedia di
sini .
Hampir semua kesalahan yang mungkin muncul selama pekerjaan dengan alat dijelaskan pada Stackoverflow atau mitra bioinformatiknya - Biostars .
Data yang digunakan
Untuk panduan, kami menggunakan data terbuka
dari proyek 1000 Genome. Untuk analisis, kami memilih 10 sampel dengan informasi tentang genotipe sekitar 85 juta variasi yang diperoleh dengan menganalisis data NGS yang selaras dengan versi genom referensi GRCh37. Hubungan keluarga dan populasi sampel ditunjukkan pada Gambar 1.
 Gambar 1
Gambar 1 Silsilah yang digunakan dalam sampel VCF. Kuadrat sesuai dengan jenis kelamin laki-laki, lingkaran ke perempuan. Garis putus-putus berarti kekerabatan urutan kedua yang tidak ditentukan.
Perhatikan
Format VCF memungkinkan Anda untuk menyimpan informasi tentang bidang seseorang sebagai nomor tunggal, jika informasi ini diketahui selama pembuatan VCF. Kelihatannya seperti ini: bidang GT (genotipe, genotipe) untuk catatan dari kromosom X berisi satu nilai numerik yang sesuai dengan satu alel, untuk pria dan dua untuk wanita. Jika tidak ada informasi tentang bidang biologis dari sampel yang diurutkan, maka bidang GT akan secara default berisi dua nilai numerik (disorot dengan warna merah pada Gambar 2).
Dalam file VCF yang digunakan dalam manual ini, kromosom Y dikeluarkan, tetapi keberadaan kromosom Y dalam file VCF tidak selalu berarti bahwa sampel yang diurutkan benar-benar memilikinya. Hal ini disebabkan oleh daerah pseudo-autosomal (PAR), yang identik untuk kromosom X dan Y dan terletak di ujungnya.
Kromosom yang berbeda biasanya tidak memiliki daerah panjang yang identik (homolog), namun kromosom X dan Y memiliki daerah tersebut beberapa juta pasangan basa panjang di awal (PAR1) dan akhir (PAR2). Oleh karena itu, ketika menganalisis data NGS pada pria di wilayah PAR, dua alel ditemukan (satu untuk setiap kromosom seks), dan pada wanita, genotipe dapat muncul di wilayah PAR dari kromosom Y, meskipun sebenarnya mereka adalah genotipe dari kromosom X mereka.
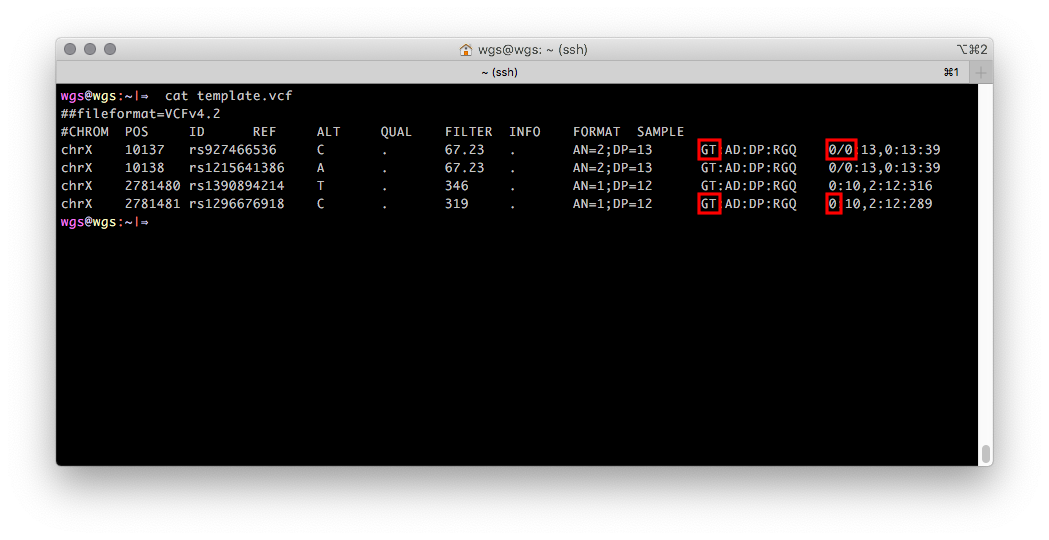 Gambar 2
Gambar 2 File VCF dengan genotipe dari kromosom X seorang pria dari wilayah PAR1 (dua entri pertama) dan wilayah non-pseudo-autosomal (dua entri terakhir).
Satuan pendidikan
Jenis kelamin genetis adalah seperangkat kromosom seks yang sesuai dengan manifestasi karakteristik seksual primer dan sekunder tipe pria atau wanita. Biasanya, pria memiliki satu kromosom X dan satu kromosom Y, sementara wanita memiliki dua kromosom X. Dengan berbagai gangguan dalam pembentukan sel kuman, telur dan sperma, seorang anak dengan seperangkat kromosom seks yang sangat baik dapat dilahirkan oleh orang tua, yang sering menyebabkan gangguan perkembangan. karakteristik seksual primer dan sekunder.
Dua anomali seksual kromosom yang paling umum adalah sindrom Turner (satu set kromosom X0, yaitu, hanya satu kromosom X) dan sindrom Klinefelter (satu set kromosom XXY).
Alel adalah satu atau lebih nukleotida yang terletak pada posisi mana pun dalam genom dan memiliki alternatif. Konsep ini digunakan untuk menggambarkan genotipe. Bedakan antara alel referensi dan alternatif. Semuanya disimpan dalam file VCF di bidang REF dan ALT, masing-masing.
Tentukan jenis kelaminnya
Untuk pengguna Yandex.CloudSemua data untuk menyelesaikan tugas-tugas manual dan independen disimpan di Yandex.Cloud menggunakan struktur yang ditunjukkan di bawah ini. Folder
Tutorial berisi file VCF yang diperlukan untuk menyelesaikan manual, folder
Test untuk tugas-tugas independen. Folder
Technical berisi dua file dengan daftar pengidentifikasi variasi genetik:
rsids_for_subsetting.txt digunakan dalam manual dan tugas-tugas untuk eksekusi independen,
external_interpretation_rsids.txt mungkin diperlukan di masa depan ketika memperoleh sekuensing lebar genom di Atlas untuk mengunggah data genotipe ke layanan pihak ketiga. Folder
Tools berisi, antara lain, dua skrip yang digunakan dalam tugas 2 dan 3.
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
Folder akan dibuat di direktori
/home di Yandex.Cloud VM, nama yang sesuai dengan nama pengguna yang ditentukan pada tahap pembuatan VM. Salin semuanya dari direktori
/home/ubuntu ke direktori Anda melalui perintah berikut:
cd ~ cp -r /home/ubuntu/* ./
Untuk sisanyaSaat mengerjakan PC pribadi, Anda dapat mengunduh file yang diperlukan untuk tugas pertama dari
tautan . Arsip yang diunduh mendukung struktur penyimpanan file yang serupa dengan yang digunakan pada Yandex.Cloud:
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
Buka arsip arsip
atlas_wgs_contest.tar.gz dengan perintah
tar -xvzf atlas_wgs_contest.tar.gz File VCF untuk melakukan tugas dalam bentuk yang tidak diarsipkan masing-masing menempati sekitar 19 gigabytes, oleh karena itu, untuk menghemat ruang, kami sarankan hanya bekerja dengan arsip. Semua program yang tercantum di atas sudah dapat bekerja dengan data VCF terkompresi. Selain itu, Anda tidak perlu melakukan apa pun.
Untuk menentukan jenis kelamin subjek, Anda perlu melihat genotipe pada kromosom X dan mengecualikan wilayah PAR1 dan PAR2 yang terletak di awal dan akhir. Ini adalah interval posisi 60001–2699520 dan 154931044–155260560 dalam versi genom GRCh37. Jika genotipe mengandung satu penunjukan numerik, ini adalah jenis kelamin biologis laki-laki, jika tidak, perempuan. Harus diingat bahwa penunjukan gender dalam file VCF tergantung pada ketersediaan informasi tentang bidang biologis selama pembuatan VCF, oleh karena itu pendekatan ini tidak selalu dapat digunakan.
Gunakan perintah berikut untuk masing-masing sampel dalam dataset. Ganti pengidentifikasi sampel setelah argumen
-s :
(/Data/Tutotrial/CEI.1kg.2019.demo.vcf.gz):
Saat menjalankan perintah, Anda akan melihat beberapa konten file VCF untuk pengidentifikasi sampel yang ditentukan.
-r chrX:2699521-154931043 di BCFtools membatasi tampilan konten file ke wilayah kromosom X dari posisi 2699521 ke posisi 154931043 (wilayah non-PAR pada Gambar 3). Batas-batas ini mengecualikan wilayah pseudo-autosomal yang tidak perlu dalam kasus ini (PAR1 dan PAR2). Menggunakan nilai numerik di bidang GT, tentukan jenis kelamin setiap sampel.
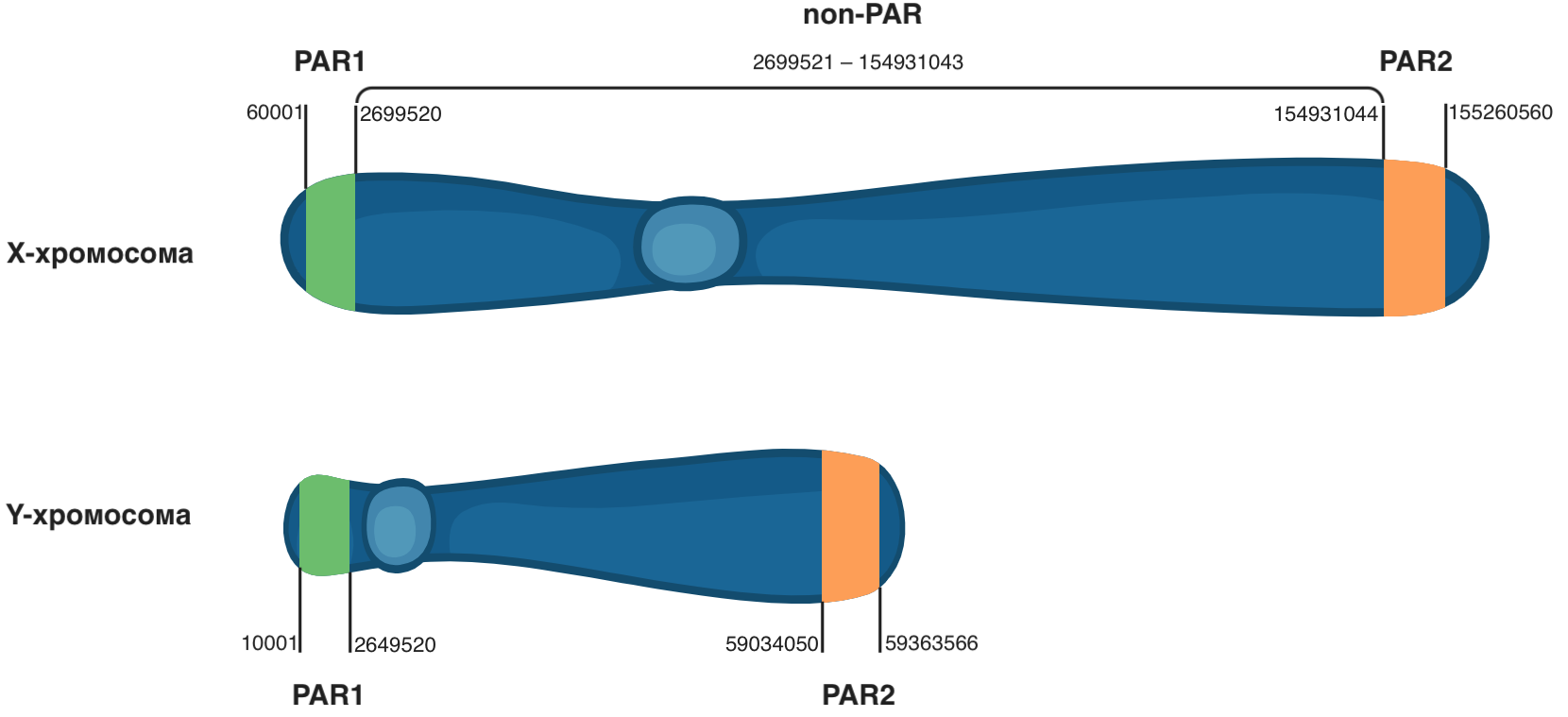 Gambar 3
Gambar 3 Lokasi pseudo-autosomal PAR1 dan PAR2 pada kromosom seks.
Anda dapat melihat daftar semua pengidentifikasi sampel dalam file VCF pada Gambar 1 atau di baris terakhir dari header file VCF. Mereka akan terdaftar setelah nama kolom FORMAT:
Jenis kelamin sebenarnya dari sampel ini juga ditunjukkan pada Gambar 1.
Kami menentukan hubungan
Untuk menentukan hubungan, kita perlu membandingkan data genetik semua sampel secara berpasangan. Sulit untuk melakukan ini sesuai dengan genom lengkap: file VCF dalam kasus ini membutuhkan puluhan gigabytes. VCF yang kami gunakan hanya memakan sekitar 2 gigabyte, tetapi kami masih memfilternya sesuai dengan daftar pengidentifikasi variasi genetik (rsIDs) yang di-genotip pada chip dari Illumina: GSA v1, GSA v2, HumanOmniExpress v1.0, HumanOmniExpress v1.3, InfiniumExome v1. 1 dan Infinium OmniExpressExome v1.4. Ini adalah chip paling populer dalam genotipe komersial.
Kami telah menyusun daftar semua pengidentifikasi variasi genetik dari chip ini dalam file terpisah dengan daftar rsIDs. Ini berisi 1,4 juta pengidentifikasi. Untuk memfilter file VCF, jalankan perintah berikut:
bcftools view -O z -i 'ID=@rsids_for_subsetting.txt' CEI.1kg.2019.demo.vcf.gz > CEI.1kg.2019.demo.subset.vcf.gz
Setiap kali Anda menggunakan BCFtools dan paket lain untuk bekerja dengan file VCF, riwayat perintah sebelumnya ditambahkan ke header file. Terlepas dari metode memfilter file VCF dan perintah yang dieksekusi sebelumnya, Anda dapat memeriksa integritas dan identitas konten utama VCF dengan menghitung jumlah hash:
Perintah
gunzip -c mendekompres file dengan output isinya ke stdout, dari mana baris header file VCF yang dimulai
# dihapus lebih lanjut (oleh karena itu digunakan perintah
grep -v "^#" ). Header dihapus untuk membandingkan integritas hanya data genetik itu sendiri, dan bukan metadata tentang alat mana dan kapan digunakan untuk bekerja dengan file VCF ini.
Jika nilai hash cocok, Anda dapat beralih dan mengonversi VCF ke format Plink internal (secara default, format Plink adalah tiga file dengan bed ekstensi, bim dan fam). Dalam file-file ini, hanya genotipe, kromosom, posisi, dan beberapa data lainnya yang tersisa, dan sisanya dihilangkan. Dengan format ini lebih mudah untuk bekerja dan menyelesaikan berbagai masalah yang tidak memerlukan informasi tambahan dari VCF. Misalnya, lakukan GWAS.
Perintah ini akan membuat tiga file di folder:
CEI.1kg.2019.demo.subset.bed
CEI.1kg.2019.demo.subset.bim
CEI.1kg.2019.demo.subset.famAnda dapat menentukan kekerabatan berpasangan untuk semua 10 sampel. Kami menggunakan perintah berikut untuk menganalisis file Plink:
king -b CEI.1kg.2019.demo.subset.bed --kinship --prefix CEI.1kg.2019.demo.subset.kinship_analysis
Lihatlah file
CEI.1kg.2019.demo.subset.kinship_analysis.kin0 dan perhatikan kolom Kekerabatan, yang berisi koefisien kekerabatan untuk pasangan sampel yang ditunjukkan masing-masing dalam ID1 dan ID2.
Bandingkan koefisien yang Anda dapatkan dalam file
CEI.1kg.2019.demo.subset.kinship_analysis.kin0 untuk semua pasangan sampel dengan silsilah yang ditunjukkan pada Gambar 1 (garis putus-putus sesuai dengan kekerabatan urutan kedua, namun, tidak ada data kekerabatan yang tepat, mis. Ini mungkin ada sepupu, bibi / keponakan atau paman / keponakan). Cobalah untuk membuat kesimpulan Anda sendiri tentang nilai-nilai koefisien kekerabatan apa yang dapat sesuai dengan keterkaitan urutan pertama dan kedua.
PetunjukKutipan dari dokumentasi KING: koefisien kekerabatan> 0,354 berhubungan dengan duplikat sampel atau kembar identik, dari 0,177 hingga 0,354 hingga kekerabatan tingkat pertama (orang tua-anak-anak, saudara kandung), dari 0,0884 hingga 0,177 hingga kekerabatan urutan kedua (sepupu, bibi, bibi) / paman-keponakan), dan dari 0,0442 hingga 0,0884 - hingga kekerabatan tingkat ketiga (kakek nenek, cucu, sepupu kedua). Apa pun yang kurang dari 0,0442 sulit untuk diartikan secara jelas.
Tugas pertama kompetisi
Menggunakan dataset uji dari 12 sampel
Data/Test/CEI.1kg.2019.test.vcf.gz ,
Data/Test/CEI.1kg.2019.test.vcf.gz silsilah mereka, dipandu oleh hasil penentuan jenis kelamin dan analisis kekerabatan. Sampel yang, menurut hasil analisis, tidak memiliki hubungan kekerabatan dengan seseorang, tulis di dekatnya, tanpa menghubungkannya dengan garis dengan sampel lain. Silsilahnya dapat dikomposisikan dalam gaya yang mirip dengan Gambar 1, namun, ini tetap pada kebijaksanaan Anda. Laki-laki ditunjukkan oleh sebuah bujur sangkar, perempuan oleh sebuah lingkaran, perkawinan dengan garis horizontal, seorang anak oleh garis vertikal, beberapa anak oleh percabangan horizontal dari garis vertikal (dalam bentuk huruf P). Baca lebih lanjut tentang penunjukan ini di
sini .
Seperti yang kami tulis di atas, koefisien kekerabatan tidak dapat dengan jelas mencirikan kekerabatan dari satu ordo lain: koefisien kekerabatan yang sama diperoleh ketika membandingkan pasangan orang tua-anak dan saudara laki-laki-perempuan (kekerabatan urutan pertama). Jika tidak mungkin untuk menetapkan sifat hubungan, tunjukkan salah satu yang mungkin. Harap dicatat bahwa sampel dalam dataset uji memiliki pengidentifikasi yang berbeda dari yang digunakan dalam dataset pelatihan.
Respons
harus dikirim ke email
wgs@atlas.ru hingga 26 Desember hingga 23:59. Dua tugas lagi akan segera diterbitkan, dan hasil akhir untuk tugas tersebut akan muncul pada 28 Desember. Pemenang akan menerima tes Genom Lengkap, dan tempat kedua dan ketiga akan menerima tes genetik Atlas. Akan ada hadiah khusus dari
Yandex.Cloud . Karyawan Atlas lama dan saat ini tidak berpartisipasi dalam kompetisi;)