Kami melanjutkan siklus tugas, di mana kami berbicara tentang cara bekerja dengan data genetik.
Tugas pertama "Cari tahu jenis kelamin dan tingkat hubungan" sudah bisa diselesaikan dan kirimkan jawaban kepada kami. Hari ini kami menerbitkan yang kedua.
Hadiah utama adalah
Genom Lengkap .

Kami sebelumnya telah membagikan informasi dan tautan bermanfaat yang mungkin berguna untuk bekerja dengan data bioinformatika. Kami menyarankan Anda membaca artikel sebelumnya terlebih dahulu jika Anda melewatkannya:
Apa genom lengkap dan mengapa itu diperlukanTugas nomor 1. Cari tahu jenis kelamin dan tingkat hubungan.Penafian
Bekerja dengan data genetik dilakukan pada sistem Unix (Linux, macOS), karena beberapa perintah dan perangkat lunak tidak tersedia di Windows. Oleh karena itu, untuk pengguna Windows, salah satu solusi paling sederhana adalah menyewa mesin virtual Linux.
Semua operasi yang dijelaskan di bawah ini dilakukan pada baris perintah - terminal. Sebelum Anda mulai, pelajari cara bekerja di terminal yang menjalankan OS Anda dan gunakan perintah, karena beberapa di antaranya berpotensi membahayakan OS dan data Anda.
Perangkat Lunak yang Diperlukan
Kami telah mengumpulkan
gambar mesin virtual (VM) dengan semua perangkat lunak yang diperlukan di Yandex.Cloud. Petunjuk untuk mengatur VM dan menginstal perangkat lunak dapat ditemukan di
artikel sebelumnya dengan Tugas No. 1.
Kali ini, Anda perlu membangun plot sebar dua dimensi menggunakan data yang diperoleh dengan metode analisis komponen utama. Kami sarankan Anda untuk membuat diagram ini menggunakan perangkat lunak apa pun yang sesuai untuk Anda: Excel, Google Sheets, Python, R, dan lainnya.
Untuk menyelesaikan tugas, Anda memerlukan paket perangkat lunak Plink 1.9. Jika Anda belum menginstalnya (dan belum menyelesaikan Tugas No. 1), baca artikel sebelumnya. Berisi instruksi instalasi. Untuk berpartisipasi dalam kompetisi Tahun Baru 2019, semua tugas harus diselesaikan!
Perhatikan
Principal component analysis (PCA) adalah salah satu algoritma pembelajaran mesin tanpa guru ketika mesin secara mandiri mencari pola dalam data. Dalam genetika, PCA memungkinkan pengelompokan sampel sesuai dengan data genotyping dalam ruang dimensi N (biasanya dua dimensi), di mana komponen utama yang diperoleh paling akurat menjelaskan variabilitas data genetik dari sampel ke sampel.
Ketika melakukan analisis seperti itu, sampel dari satu populasi biasanya membentuk sebuah cluster, ukuran dan kehalusan batas yang bergantung pada kesamaan sampel dalam populasi tertentu. Algoritma ini kemungkinan untuk mengidentifikasi sampel dari populasi yang berbeda dalam kelompok yang berbeda. Dan sampel dari populasi dekat yang termasuk dalam superpopulasi yang sama, misalnya, EAS - Asia Timur, seperti pada Gambar 1, akan diidentifikasi secara dekat satu sama lain atau bahkan dalam cluster yang berpotongan.
 Gambar 1
Gambar 1 Silsilah sampel yang digunakan dalam VCF (kuadrat sesuai dengan jenis kelamin laki-laki, lingkaran ke perempuan). Garis putus-putus sesuai dengan hubungan urutan kedua yang tidak ditentukan.
Analisis serupa digunakan untuk menentukan populasi dengan genotipe. Untuk ini, diperlukan dataset referensi, yang terdiri dari sampel dengan asal yang sudah diketahui. Kesimpulan tentang populasi dapat dibuat oleh kelompok sampel mana yang diketahui paling dekat dengan data yang diteliti.
Untuk menyederhanakan, esensi dari analisis PCA adalah bahwa jarak berpasangan antara titik-titik dalam ruang multidimensi diketahui, dan titik-titik ini harus ditempatkan dalam ruang dimensi yang lebih kecil sehingga jarak berpasangan yang baru minimal berbeda dari yang asli. Reduksi dimensi menyederhanakan analisis data, tetapi semakin kita menguranginya, semakin kuat jarak baru antara titik-titik berbeda dari aslinya. Oleh karena itu, tugas analisis PCA juga melibatkan menemukan kompromi antara akurasi dan kemudahan analisis. Semuanya seperti dalam hidup.
Perwujudan paling sederhana dari implementasi PCA pada data genetik didasarkan pada identitas beberapa alel, yang dapat dibagi menjadi dua subtipe: IBS (identitas berdasarkan negara) dan IBD (identitas berdasarkan keturunan). IBS berarti identitas alel tertentu dalam dua orang, tetapi tidak selalu menyiratkan fakta adanya hubungan di antara mereka. IBD, sebaliknya, berbicara tentang identitas alel karena keberadaan leluhur yang sama dan, karenanya, kekerabatan.
Alel IBD adalah alel IBS yang jelas, sedangkan yang sebaliknya tidak benar. Namun, harus diingat bahwa pada suatu saat kita berasal dari nenek moyang yang sama, sehingga beberapa alel mungkin IBD. Dalam analisis PCA di bawah ini, hanya konsep IBS yang digunakan, meskipun dalam analisis yang lebih kompleks itu memperhitungkan tes signifikansi statistik, batasan fenotipik, ukuran cluster, usia dan jenis kelamin orang tersebut, serta informasi tambahan tentang struktur populasi.
Semakin besar jumlah alel yang berbeda dalam dua sampel, semakin sedikit mereka mirip dan semakin jauh mereka dari satu sama lain. Nilai IBS untuk sampel tersebut akan rendah. Tetapi untuk orang tua dan anak-anak mereka, IBS akan sangat tinggi.
Mengetahui nilai-nilai IBS untuk setiap pasangan gambar dalam dataset, Anda dapat melakukan analisis PCA untuk melihat bagaimana mereka berkerumun.
Tes genetik Atlas menggunakan algoritma yang jauh lebih canggih untuk menentukan perwakilan populasi dalam data genotipe.
Data yang digunakan
Kami mengingatkan Anda bahwa manual ini menggunakan data terbuka yang dipilih secara khusus dari proyek
1000 Genome . Untuk analisis, dipilih 10 sampel dengan informasi genotipe ~ 85 juta variasi, yang diperoleh dengan menganalisis data NGS yang selaras dengan versi genom GRCh37. Hubungan keluarga dan populasi sampel ini ditunjukkan pada Gambar 1.
Membangun kelompok populasi
Gunakan tiga file dalam format Plink yang diperoleh sebelumnya di Tugas No. 1:
CEI.1kg.2019.demo.subset.bed CEI.1kg.2019.demo.subset.bim CEI.1kg.2019.demo.subset.fam
Tentukan jarak berpasangan antara semua 10 sampel dalam dataset pelatihan dan gambar PCA berdasarkan IBS (identitas berdasarkan negara). Ini dapat dilakukan sebagai berikut:
Parameter
—genome hanya bertanggung jawab untuk perhitungan berpasangan IBS / IBD antara semua sampel dalam dataset. Parameter "
—read-genome " adalah matriks jarak berpasangan yang diperoleh sebelumnya, dan parameter "
—cluster —mds-plot 10 bertanggung jawab untuk analisis PCA dan output dari hasilnya ke tabel 10 komponen utama pertama. Bahkan, ini adalah koordinat masing-masing sampel dalam ruang 10-dimensi.
Perintah terakhir akan membuat 4 file di folder:
CEI.1kg.2019.demo.subset.clustering.cluster1 CEI.1kg.2019.demo.subset.clustering.cluster2 CEI.1kg.2019.demo.subset.clustering.cluster3 CEI.1kg.2019.demo.subset.clustering.mds
Kami akan membutuhkan dua file terakhir dari daftar.
Gambar 2 menunjukkan bagaimana file yang diterima pada dataset pelatihan MDS terlihat. Bidang FID (Family ID) dan IID (Individual ID) sesuai dengan keluarga dan pengidentifikasi sampel individu. Fields C1 - C10 berisi nilai masing-masing dari sepuluh komponen utama untuk setiap sampel, di mana komponen C1 secara maksimal menjelaskan variabilitas data genetik sampel yang dianalisis, dan C10 minimal.
 Gambar 2
Gambar 2 File MDS dengan nilai 10 komponen utama untuk setiap sampel.
Ketika membangun diagram sebar menggunakan dua komponen (dalam ruang dua dimensi), seseorang dapat mendeteksi cluster yang sesuai dengan populasi sampel. Gambar 3 menunjukkan plot pencar untuk pasangan komponen utama C1xC2, C2xC3 dan C1xC3. Ketika membandingkan cluster yang diperoleh dengan afiliasi populasi referensi (Gambar 1), pasangan dari dua komponen pertama C1 - C2 menunjukkan akurasi tertinggi (100%), dengan benar memisahkan semua sampel sesuai dengan afiliasi populasi mereka yang dinyatakan dalam proyek 1000 Genom. Namun, selalu masuk akal untuk membandingkan hasil yang diperoleh untuk beberapa pasang komponen karena kemungkinan tumpang tindih atau pemisahan cluster nyata.
 Gambar 3
Gambar 3 Plot pencar lokasi sampel untuk pasangan komponen utama; lokasi marker sedikit diubah untuk mencegah mereka tumpang tindih.
Membangun diagram 3D menggunakan tiga komponen utama pertama juga dapat membantu menentukan pengelompokan, tetapi tidak selalu. Sebagai contoh, membangun diagram seperti itu untuk data pada Gambar 3 memungkinkan kita untuk mengidentifikasi 4 cluster di mana sampel dari populasi PUR dan KHV dikelompokkan menurut populasi, dan sampel dari populasi CDX dibagi menjadi dua kelompok (Gambar 4). Ini juga terlihat pada Gambar 3 dalam koordinat C2xC3 dan C1xC3.
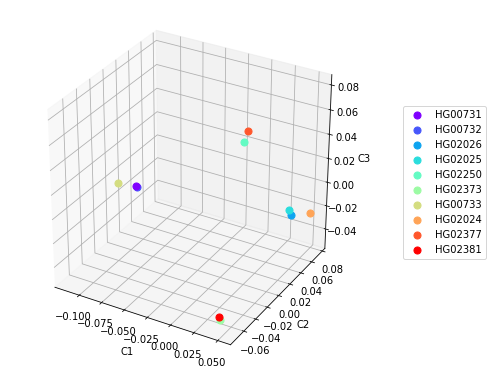 Gambar 4
Gambar 4 Plot pencar untuk tiga komponen utama.
Hasil analisis yang saling bertentangan tersebut dapat dijelaskan oleh sejumlah kecil sampel, karena nilai-nilai komponen utama dari masing-masing sampel berbeda untuk ukuran dan komposisi data yang berbeda, dan ketika sampel tambahan dari populasi yang berbeda dimasukkan, hasil pengelompokan dapat berubah. Kesalahan juga berpotensi terjadi saat membuat dataset dan memberikan data referensi tentang populasi sampel, namun, dalam proyek 1000 Genom, kemungkinan situasi seperti itu cukup rendah.
File MDS tidak menggunakan tab atau koma sebagai pembatas, jadi sesuaikan formatnya untuk kenyamanan. Gunakan
tab atau
csv sebagai argumen kedua:
Tim akan membuat file
CEI.1kg.2019.demo.subset.clustering.mds.tab , yang dapat Anda unduh dan buat bagan
CEI.1kg.2019.demo.subset.clustering.mds.tab yang mirip dengan yang ditunjukkan pada Gambar 3. Bandingkan hasilnya, mereka harus identik dengan yang ditunjukkan di atas.
Membangun pohon pengelompokan
Anda juga dapat mengevaluasi pengelompokan sampel menggunakan pohon biner, yang mewakili informasi pengelompokan tentang sampel dalam bentuk diskrit. Informasi tentang pohon ini terkandung dalam file
CEI.1kg.2019.demo.subset.clustering.cluster3 (Gambar 5).
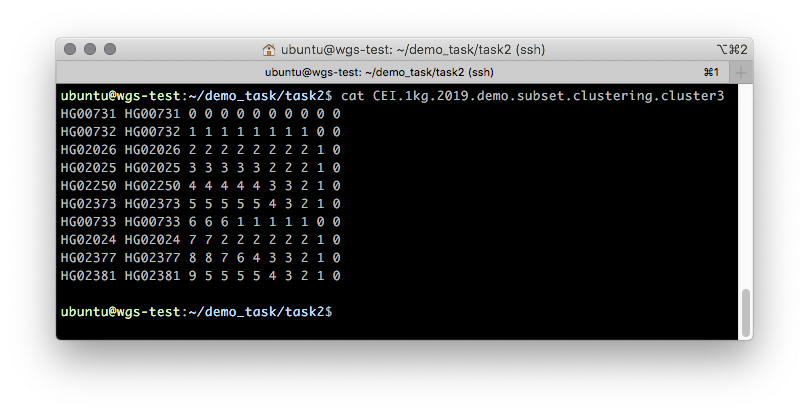 Gambar 5
Gambar 5 Perkiraan konten file
.cluster3 yang menjelaskan proses pengelompokan sampel bertahap dari 1 kluster ke N, di mana N adalah jumlah sampel.
Dua kolom pertama dari file ini berisi FID dan IID. Afiliasi cluster dijelaskan oleh semua orang. File ini harus dibaca dari kanan ke kiri dalam kolom dengan penambahan satu kolom: pada awalnya, semua sampel milik satu kluster “0” - kolom paling kanan. Ketika dibagi menjadi dua kluster (pada langkah kedua, pada kolom kedua), dua kluster muncul: “0” dan “1”, di mana kluster “0” berisi sampel HG00731, HG00732 dan HG00733, dan kluster “1” berisi sisanya. Ilustrasi partisi seperti itu ditunjukkan pada Gambar 6.
Dari pohon, dapat disimpulkan bahwa sampel milik populasi (Gambar 1). Selain itu, konstruksi pohon ini memungkinkan kita untuk menentukan kedekatan populasi individu, yaitu, terjadinya populasi CDX dan KHV dalam satu superpopulasi EAS (sudah pada langkah pertama pemisahan superpopulasi EAS dan AMR dipisahkan dalam dua cabang yang ada). Juga, konstruksi pohon pengelompokan dapat membantu untuk memperbaiki hasil ambigu dari visualisasi sampel pada komponen utama.
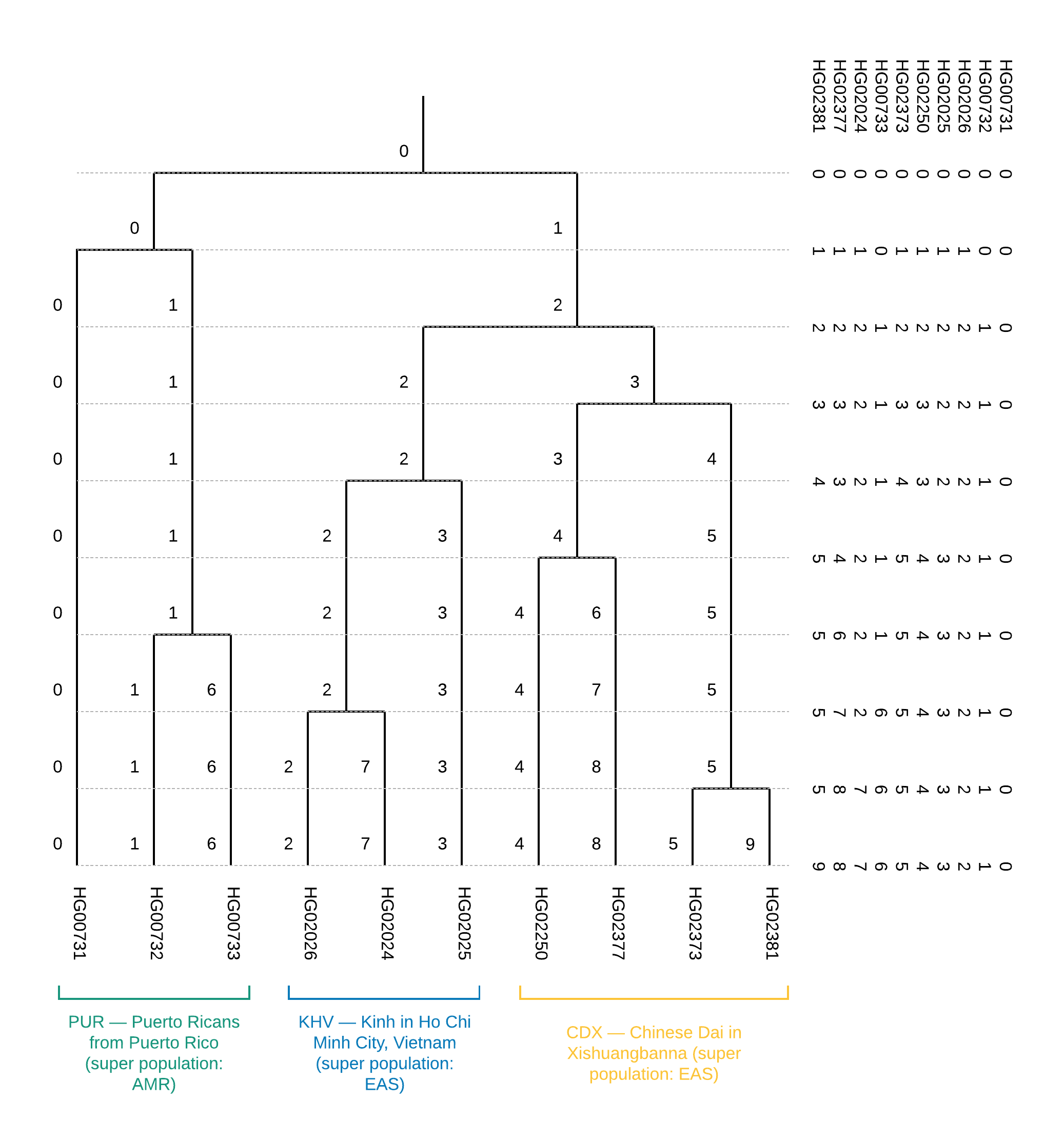 Gambar 6
Gambar 6 Binary clustering tree untuk dataset pelatihan 10 sampel: di sebelah kanan adalah isi dari file
CEI.1kg.2019.demo.subset.clustering.cluster3 (dari kanan ke kiri dalam file, identik dari atas ke bawah pada gambar).
Tugas kedua dari kompetisi
Gunakan dataset uji dari 12 sampel
Data/Test/CEI.1kg.2019.test.vcf.gz dan contoh di atas (Gambar 5) untuk membangun pohon pengelompokan biner dari file
.cluster3 Anda
.cluster3 dan lampirkan pada solusi. Menganalisis pohon yang dihasilkan dan menarik kesimpulan tentang jumlah populasi super yang disajikan dalam dataset uji.
Tentukan pengelompokan populasi dari 12 sampel dari dataset uji dengan menganalisis komponen utama C1, C2 dan C3 dengan mempertimbangkan pohon yang dibangun dan mengindikasikan ini pada silsilah yang dibangun pada Masalah No. 1, membatasi blok populasi individu (mirip dengan Gambar 1). Sampel yang tidak menunjukkan adanya kekerabatan dalam Soal No. 1 harus ditempatkan dengan cara yang sama di dalam blok yang diperoleh dalam diagram tanpa menghubungkannya dengan garis dengan sampel lain. Jangan lupa lampirkan plot pencar yang Anda buat.
Respons
harus dikirim ke email
wgs@atlas.ru hingga 26 Desember hingga 23:59. Tugas lain akan segera diterbitkan, dan hasil akhir untuk tugas akan muncul pada 28 Desember. Pemenang akan menerima tes Genom Lengkap, dan tempat kedua dan ketiga akan menerima tes genetik Atlas. Akan ada hadiah khusus dari
Yandex.Cloud . Karyawan Atlas lama dan saat ini tidak berpartisipasi dalam kompetisi;)