Hari ini kami menerbitkan tugas terakhir dari siklus di mana kami memberi tahu cara bekerja dengan data genetik.
Tugas pertama dan
kedua sudah diterbitkan: mereka dapat diselesaikan dan mengirim jawaban kepada kami. Kami memperingatkan Anda bahwa tugas ini lebih lama dari yang lain.
Hadiah utama adalah
Genom Lengkap .

Kami sebelumnya telah membagikan informasi dan tautan bermanfaat yang mungkin berguna untuk bekerja dengan data bioinformatika. Kami menyarankan Anda membaca artikel sebelumnya terlebih dahulu jika Anda melewatkannya:
Apa genom lengkap dan mengapa itu diperlukanTugas nomor 1. Cari tahu jenis kelamin dan tingkat hubungan.Tugas nomor 2. Penentuan struktur populasiPenafianBekerja dengan data genetik dilakukan pada sistem Unix (Linux, macOS), karena beberapa perintah dan perangkat lunak tidak tersedia di Windows. Oleh karena itu, untuk pengguna Windows, salah satu solusi paling sederhana adalah menyewa mesin virtual Linux.
Semua operasi yang dijelaskan di bawah ini dilakukan pada baris perintah - terminal. Sebelum Anda mulai, pelajari cara bekerja di terminal yang menjalankan OS Anda dan gunakan perintah, karena beberapa di antaranya berpotensi membahayakan OS dan data Anda.
Perangkat Lunak yang Diperlukan
Kami telah mengumpulkan
gambar mesin virtual (VM) dengan semua perangkat lunak yang diperlukan di Yandex.Cloud. Petunjuk untuk menyiapkan VM dan menginstal perangkat lunak dapat ditemukan di
artikel dengan tugas pertama. Ada juga instruksi tentang cara mengatur mesin untuk menggunakannya secara gratis hingga 31 Desember 2019.
Dalam tugas ini, Anda perlu mengonversi data genotip dari format VCF ke format 23andMe, mengunggah file yang diterima ke layanan Promethease dan berkenalan dengan isi laporan untuk setiap sampel.
Format 23andMe adalah format teks untuk menyimpan data genotyping dan berisi 4 bidang yang dipisahkan oleh tab. Bidang pertama berisi pengidentifikasi variasi (misalnya, rsID), bidang kedua berisi kromosom (nilai yang valid untuk bidang ini adalah 1-22, X, Y, dan MT), bidang ketiga berisi posisi pada kromosom, yang keempat berisi genotipe (diploid di hadapan dua kromosom homolog, haploid di tempat lain) kasus). Format ini didukung oleh banyak layanan interpretasi, jadi dalam tugas kami akan bekerja dengannya.
Untuk menyelesaikan tugas, Anda memerlukan paket perangkat lunak BCFtools. Jika Anda belum menginstalnya, baca
artikel dengan tugas pertama. Berisi instruksi instalasi. Kami mengingatkan Anda bahwa untuk berpartisipasi dalam kompetisi Tahun Baru 2019, semua tugas harus diselesaikan.
Selain BCFtools, Anda harus membuat
create_23andme.sh - skrip bash yang digunakan untuk menghasilkan data dalam format 23andMe. File ini terletak di direktori
/Technical di Yandex.Cloud serta di arsip untuk diunduh, tersedia melalui tautan di
artikel .
Perhatikan
Ada banyak layanan yang menganalisis data genotipe: MyHeritage, Promethease, FamilyTreeDNA, DNA.LAND, GEDmatch. Mereka menyediakan unduhan data genotip dalam berbagai format, seringkali khusus untuk penyedia genotip tertentu (Ancestry, 23andMe, MyHeritage, FamilyTreeDNA, GenesForGood, dan lainnya). Yang paling setia pada format data adalah Promethease: Anda dapat mengunduh file VCF dan 23andMe ke dalam layanan ini.
Ada beberapa masalah kompatibilitas antara format dan layanan:
- Perusahaan yang berbeda menggunakan versi genom yang berbeda untuk memetakan variasi genetik. Masalah ini diselesaikan dengan apa yang disebut liftover, ketika posisi variasi genetik dalam sumber data digantikan oleh yang sesuai dalam versi genom yang lain. Misalnya, Atlas menyediakan data genotipe untuk versi genom GRCh38, dan GEDmatch menerima data untuk versi genom GRCh37 sebelumnya. Konversi koordinat variasi genetik dari GRCh38 ke GRCh37 disebut lift.
- Menggunakan pengidentifikasi unik untuk variasi genetik selain rsIDs. Ketidakcocokan tersebut diselesaikan dengan mengecualikan entri seperti itu dari file atau dengan membubuhi keterangan dengan menetapkan rsID. Yang kedua tidak selalu memungkinkan.
- Layanan menggunakan serangkaian variasi genetik yang tetap. Terkadang ketidakcocokan setidaknya sebagian data yang diunduh menghasilkan kesalahan pemuatan. Masalah ini relevan, misalnya, untuk MyHeritage. Ini dapat diselesaikan dengan menyorot satu set pengidentifikasi variasi genetik yang tidak menyebabkan kesalahan pemuatan.
Data yang digunakan
Kami mengingatkan Anda bahwa manual ini menggunakan data terbuka yang dipilih secara khusus dari proyek
1000 Genome . Untuk analisis, kami memilih 10 sampel dengan informasi genotipe ~ 85 juta variasi, yang diperoleh dengan menganalisis data NGS yang selaras dengan versi genom GRCh37. Hubungan keluarga dan populasi sampel ini ditunjukkan pada Gambar 1.
 Gambar 1
Gambar 1 Silsilah sampel yang digunakan dalam VCF (kuadrat sesuai dengan jenis kelamin laki-laki, lingkaran ke perempuan). Garis putus-putus sesuai dengan hubungan urutan kedua yang tidak ditentukan.
Konversi VCF
Di bawah ini adalah petunjuk untuk mengonversi file VCF dan mengunggah data yang diterima ke layanan Promethease, yang baru-baru ini menjadi gratis. Kami menyarankan Anda membiasakan diri dengan laporan Promethease yang diterima pada sampel mana pun. Gunakan file VCF yang difilter menurut daftar variasi yang diperoleh pada
tugas No. 1 .
bcftools query memungkinkan Anda untuk mengekstrak informasi yang tersedia dalam format yang ditentukan oleh pengguna setelah flag
-f dari file VCF. Bendera
-s menunjukkan pengidentifikasi sampel (
HG00731 ) untuk mengekstraksi data. Bendera -e digunakan untuk menunjukkan kriteria pengecualian, dalam hal ini
'%ID=="."' Tidak termasuk entri yang tidak memiliki rsID. Output dari
bcftools query diteruskan ke script
create_23andme.sh , yang mengubah data ke format TSV dengan 4 kolom (rsID, kromosom, posisi, genotipe) dan menulisnya ke file. Anda dapat mengunduh dan menyimpan skrip
create_23andme.sh Anda dapat bekerja dengan data sekuensing genom lengkap.
Skrip
create_23andme.sh menggunakan
create_23andme.sh diekstraksi dari file VCF untuk menentukan jenis variasi genetik (variasi nukleotida tunggal SNV, penyisipan INS atau penghapusan DEL) dan menulis pengidentifikasi rsID, kromosom, posisi dan alel dalam
stdout sesuai dengan jenis variasi spesifik (A, G, T, T dan C adalah alel yang valid untuk tipe SNV, I dan D adalah sebutan alel yang valid untuk tipe INS dan DEL).
Ingatlah bahwa proses konversi membutuhkan banyak waktu: sekitar 4 jam per file untuk satu sampel dengan ~ 1 juta variasi. Concurrency BCFtools tidak didukung.
Pergi ke
promethease.com dan daftar. Klik tombol Unggah data mentah (Gambar 2) dan unggah file
HG00731.subset.23andme.txt . Setelah unduhan selesai, klik tombol Buat laporan gratis dan masukkan nama laporan yang diinginkan yang akan dihasilkan sesuai dengan data Anda. Setelah menyusun laporan, Anda akan menerima pemberitahuan melalui email dan Anda dapat membiasakan diri dengan isi laporan. Dalam laporan untuk setiap sampel, temukan golongan darah yang ditentukan oleh sistem interpretasi Promethease dalam sistem AB0 / Rh (faktor Rh-Rh). Periksa hasil Anda untuk kepatuhan dengan Tabel 1.
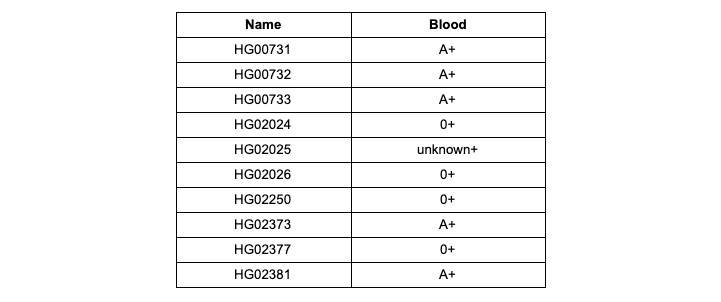 Tabel 1
Tabel 1 . Golongan darah dan faktor Rh diperoleh dari analisis sampel Promethease dari dataset demo
Atlas menggunakan ambang batas yang berbeda dari Promethease untuk memasukkan atribut tertentu dalam interpretasi berdasarkan tingkat bukti. Tingkat bukti mengacu pada totalitas hasil uji statistik dan kriteria untuk signifikansi setiap hubungan yang diamati antara variasi genetik dan fitur apa pun dari tubuh manusia. Banyak sifat yang dapat ditemukan dalam laporan Promethease memiliki tingkat bukti yang rendah dan / atau tingkat tinggi hanya dalam kumpulan populasi yang terbatas, misalnya, hanya untuk perwakilan populasi Asia.
CatatanSecara empiris, kami telah memasang daftar variasi genetik berdasarkan chip
Infinium Global Screening Array v2.0 yang dapat diunggah ke MyHeritage. Daftar ini (
external_interpretation_rsids.txt ) disimpan dalam file terpisah di direktori
/Technical , dan dapat digunakan untuk memfilter VCF dengan konversi selanjutnya dengan analogi dengan instruksi di atas. Anda juga dapat menggunakan file ini untuk memfilter data genotyping dari sebuah chip sehingga Anda dapat mengunggahnya ke MyHeritage. Jika Anda sudah memiliki tes genetik Atlas, Anda dapat mengunggah data genotyping dalam format dari akun pribadi Anda dan memfilternya sesuai dengan daftar variasi yang diusulkan - kolom pertama dalam data yang diunggah dari akun pribadi Anda.
Perhatikan bahwa file yang digunakan dalam manual ini selalu berisi bidang ALT yang diisi (alel alternatif), yang memungkinkan untuk memahami jenis apa yang dimiliki setiap variasi (INS, DEL, SNV) dan dengan benar membuat entri dalam format 23andMe. Data sekuensing genom-lebar dalam Atlas berisi alel ALT yang diisi hanya di tempat-tempat di mana alel ini terdeteksi, jika tidak informasi untuk mengisi bidang ALT saat membuat file VCF sama sekali tidak ada. Output data di situs referensi homozigot (posisi dalam genom di mana alel referensi tidak ditemukan) diperlukan, karena tidak hanya variasi yang terdeteksi dalam urutan nukleotida memiliki efek klinis, tetapi juga tidak adanya mereka.
Tidak adanya alel ALT pada posisi genom seperti itu tidak memungkinkan kita untuk menentukan jenis variasi genetik yang hanya ditemukan alel referensi (REF). Merekam genotipe untuk kasus-kasus seperti itu rumit oleh kebutuhan untuk menggunakan sumber informasi tentang kemungkinan alel untuk variasi ini dan tidak dicakup oleh panduan ini. Jika Anda berpotensi menggunakan manual ini dan skrip
create_23andme.sh untuk mengonversi file VCF yang diperoleh setelah sekuensing lebar genom ke Atlas, file yang dikonversi tidak akan berisi referensi genotipe homozigot, karena skrip
create_23andme.sh secara eksplisit menyaring catatan tersebut untuk menghilangkan kesalahan saat membuat catatan untuk penyisipan dan penghapusan.
Agar skrip
create_23andme.sh tetap menghasilkan referensi genotipe homozigot, Anda perlu mengganti konten baris 25–28 di dalamnya
... if [ "$ALT" == "." ] || [[ "$ALT" == *"*"* ]] then continue fi ...
pada
... if [[ "$ALT" == *"*"* ]] then continue fi if [ "$ALT" == "." ] then echo -e "$RSID\t$CHR\t$POS\t$REF$REF" fi ...
Substitusi ini memungkinkan entri
stdout dengan genotipe referensi homozigot untuk ditampilkan. Harus diingat bahwa entri untuk penyisipan dan penghapusan akan salah, karena alel yang valid dalam format yang digunakan untuk penyisipan dan penghapusan adalah I dan D, dan skrip akan menggunakan alel A, G, T, atau C. Untuk menghasilkan data dengan benar insersi dan penghapusan, perlu diketahui sebelumnya tentang jenis variasi apa yang menjadi ciri khas posisi genom tertentu di mana alel ALT tidak terdeteksi. Informasi ini dapat diperoleh dengan menganalisis alel ALT jika tersedia (sudah diterapkan di
create_23andme.sh ) atau menggunakan database eksternal, misalnya, dbSNP (bukan di
create_23andme.sh ).
Untuk mendapatkan laporan Promethease pada file VCF lengkap dari sekuensing genom penuh di Atlas, Anda dapat memuat file VCF itu sendiri ke dalam Promethease, namun, Anda perlu diingat bahwa ukuran file Atlas VCF yang dikompresi adalah sekitar 8 gigabyte, sementara Promethease memungkinkan Anda untuk mengunggah file tidak lebih dari 4 gigabytes. Deskripsi solusi untuk masalah ini tersedia di
sini . Solusi lain adalah dengan membagi file VCF menjadi beberapa bagian (masing-masing kurang dari 4 gigabytes) dan memuat masing-masing sebagai file tambahan dalam menu pengunduhan data Promethease.
Tugas ketiga kompetisi
Unduh data yang dikonversi dari masing-masing 12 sampel dataset uji, yang Anda filter sesuai dengan daftar variasi dalam tugas pertama, di Promethease dan kompilasi tabel korespondensi untuk pengidentifikasi sampel - golongan darah AB0 / Rh yang ditentukan oleh sistem interpretasi Promethease (faktor Rh-Rh). Golongan darah diidentifikasi secara probabilistik dan dicatat dengan awalan "prob" dalam laporan Promethease, tulis tanpa awalan. Catat nilai yang tidak terdefinisi sebagai tidak diketahui (Faktor rhesus untuk golongan darah yang tidak diketahui masih perlu ditulis, jika didefinisikan). Contoh disajikan pada Tabel 1.
Mengubah VCF ke format yang digunakan di atas dalam implementasi yang diusulkan sangat disederhanakan, tetapi membutuhkan sejumlah waktu yang signifikan. Untuk optimisasi, Anda dapat menulis skrip dengan loop yang akan secara otomatis menghasilkan data ini, iterasi pada set pengidentifikasi. Dimungkinkan untuk membuat beberapa skrip tersebut dan untuk setiap transfer set pengidentifikasi sampel yang berbeda untuk eksekusi paralel, namun, jumlah skrip yang berjalan paralel tidak boleh melebihi jumlah CPU komputer / mesin virtual Anda. Deskripsi yang baik untuk membuat loop seperti itu tersedia di
sini . Saat mengerjakan Yandex.Cloud, Anda dapat, jika perlu, membuat mesin virtual lain dengan sejumlah besar CPU virtual, yang secara proporsional akan mengurangi waktu yang dibutuhkan untuk menyelesaikan tugas.
Ini adalah tugas terakhir dari siklus kami. Respons
harus dikirim ke email
wgs@atlas.ru hingga 26 Desember hingga 23:59. Kami akan mempublikasikan jawaban yang benar dan nama-nama pemenang pada 28 Desember. Pemenang akan menerima tes Genom Lengkap, dan tempat kedua dan ketiga akan menerima tes genetik Atlas. Akan ada hadiah khusus dari
Yandex.Cloud . Karyawan Atlas lama dan saat ini tidak berpartisipasi dalam kompetisi;)