Quando o morfologista britânico George Jackson Mywart [St. George Jackson Mivart] publicou em 1865 uma das primeiras árvores evolutivas; ele carecia de material de apoio. Ele construiu uma árvore - um
mapa de várias espécies de primatas - usando uma análise detalhada de espinhos de animais. A segunda árvore, baseada na comparação de membros de animais,
mostrou outros laços familiares entre primatas, destacando o problema da biologia evolutiva que existe até hoje.
Quase 150 anos depois, os cientistas adquiriram montanhas de dados para construir as chamadas
árvores filogenéticas , uma versão moderna da estrutura criada por Mivart. Os avanços na tecnologia de decodificação de DNA e na
bioinformática permitem comparar as seqüências de centenas de genes e, às vezes, genomas inteiros, de diferentes espécies, e criar a árvore da vida com mais detalhes do que nunca.
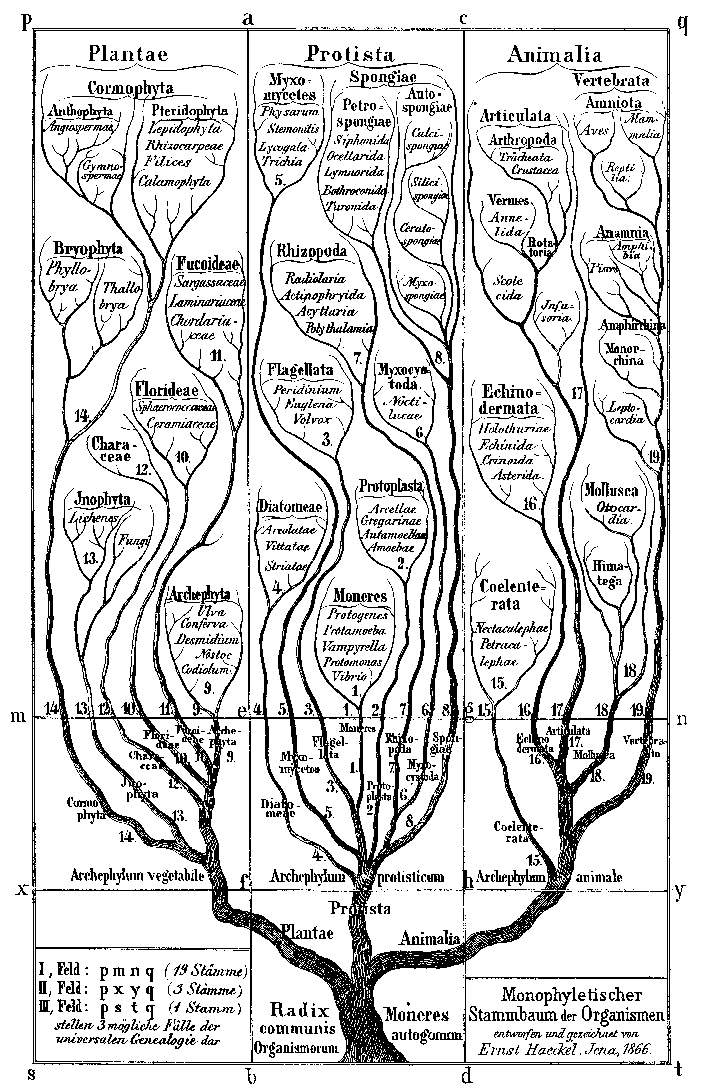 A histórica árvore da vida de 1866 descreve os reinos de plantas, animais e animais unicelulares.
A histórica árvore da vida de 1866 descreve os reinos de plantas, animais e animais unicelulares.Mas, embora a abundância de dados tenha ajudado a resolver alguns dos conflitos que surgiram em diferentes partes da árvore evolutiva, também trouxe novas dificuldades. A versão atual da árvore da vida é mais uma página controversa da Wikipedia do que um livro publicado - há debates em andamento sobre alguns ramos. Assim como a coluna vertebral e os membros levaram ao surgimento de mapas conflitantes da evolução dos primatas, os cientistas agora sabem que genes diferentes no mesmo corpo podem contar histórias diferentes.
De acordo com um novo estudo, parcialmente baseado no estudo de leveduras, o quadro controverso desenhado por genes individuais é ainda mais contraditório do que o esperado. "Alega-se que cada um dos 1070 genes está envolvido em algum tipo de conflito", diz Michael
Donoghue , biólogo evolutivo de Yale que não está associado ao estudo. "Estamos tentando descobrir as relações filogenéticas de 1,8 milhão de espécies e não podemos separar vinte tipos de leveduras", diz ele.
Para resolver o paradoxo, os pesquisadores desenvolveram um algoritmo baseado na teoria da informação para medir o nível de confiança na correção de partes individuais da árvore. Eles esperam que a nova abordagem ajude a esclarecer os períodos de evolução que possuem os dados mais interessantes e úteis e os mais conflitantes - por exemplo, a
explosão cambriana - a rápida diversificação da vida animal que ocorreu 540 milhões de anos atrás.
"Historicamente, os episódios mais interessantes estão relacionados a áreas que atraíram a atenção e causaram polêmica", como a origem de animais, vertebrados e plantas com flores, diz
Antonis Rokas , biólogo da Universidade Vanderbilt, que liderou o novo estudo.
Com base nos resultados do novo algoritmo, os cientistas podem selecionar apenas os genes mais informativos para a construção de árvores filogenéticas. Essa abordagem pode tornar o processo mais preciso e eficiente. "Acho que isso ajudará a acelerar a reconstrução da árvore da vida", disse Khidir Hilu, biólogo do Instituto de Tecnologia da Virgínia.
Tijolos da vida
A base das árvores filogenéticas é criada através do agrupamento de espécies de acordo com seu grau de parentesco. Se compararmos o DNA de humanos, chimpanzés e peixes, fica claro que humanos e chimpanzés estão mais próximos um do outro do que os peixes.
Era uma vez, os pesquisadores usavam um ou mais genes para comparar organismos. Mas, na última década, houve uma explosão de dados filogenéticos, preenchendo rapidamente as bases necessárias para criar essas árvores. A análise preencheu vários pontos brancos espalhados pela árvore, mas ainda existem discordâncias sérias.
Por exemplo, ainda não está claro quem é o mais próximo dos caracóis - moluscos bivalves ou moluscos
com patas de pá , diz Rokas. Não se sabe exatamente como alguns dos primeiros ramos de animais de uma árvore, como água-viva e esponjas, estão interconectados. Os cientistas podem mostrar exemplos de árvores conflitantes que aparecem nos mesmos periódicos científicos com uma diferença de semanas ou
mesmo no mesmo número .
"Daí a pergunta: por que é tão difícil concordar?" - diz Rokas.

Rokas e seu aluno de pós-graduação Leonidas Salichos [Leonidas Salichos] estudaram essa questão,
avaliando genes individualmente , usando os genes mais úteis - transferindo a maioria das informações relacionadas à história da evolução - para construir sua versão da árvore.
Eles começaram com 23 espécies de leveduras e selecionaram 1.070 genes. Para começar, eles criaram uma árvore filogenética de uma maneira padrão, a concatenação. Para fazer isso, todas as seqüências de espécies individuais são reunidas em um megágeno e, em seguida, as seqüências de espécies individuais são comparadas com essa longa sequência, com base na qual é criada uma árvore que melhor explica as diferenças.
A árvore resultante é precisa em termos de análise estatística padrão. Porém, como métodos semelhantes levam a árvores cheias de desacordo, Rokas e Salichos decidiram se aprofundar no assunto. Eles construíram conjuntos de árvores filogenéticas para genes de leveduras individuais e aplicaram um algoritmo desenvolvido usando a teoria da informação para procurar áreas de maior correspondência entre diferentes árvores. O resultado,
publicado na revista Nature em maio , foi inesperado. Cada gene estudado parece contar uma história evolutiva ligeiramente diferente.
"Quase todas as árvores construídas para genes individuais colidiram com uma árvore com base na concatenação de dados", diz Hilu. "Isso é chocante."
Eles concluíram que, se vários genes suportam uma arquitetura específica, ela deve ser precisa. Mas se conjuntos diferentes de genes suportam igualmente duas arquiteturas diferentes, a probabilidade de sua correspondência exata com a realidade é reduzida. Rokas e Salichos usaram um método chamado
bootstrap estatístico para selecionar os genes mais informativos.
De fato, "se você pegar apenas genes com suporte ativo, obterá a árvore certa", diz Donogue.
A árvore revisada coincidiu com uma árvore construída sobre uma fonte alternativa de informação evolutiva - mudanças em larga escala nos segmentos de DNA transmitidos de geração em geração - que justificaram suas pesquisas.
As descobertas não se limitaram ao fermento. Aplicando a mesma análise a formas de vida maiores e mais complexas, incluindo dados genéticos de vertebrados e animais, eles encontraram sérios conflitos entre genes individuais.
Alguns pesquisadores precisam se acostumar com a idéia de excluir seletivamente os dados da análise. "Por muitos anos, o principal problema para as pessoas que tentam entender as relações dos organismos tem sido o problema de coletar dados suficientes", diz
Jeffrey Townsend , biólogo evolucionário de Yale que não está relacionado à pesquisa. "A comunidade sempre foi informada sobre a necessidade de um conjunto de dados, portanto, não é surpreendente que eles abordem a tarefa dessa maneira."
Embora os biólogos evolucionistas tenham lutado com esses problemas há anos, o novo estudo se tornou a maior tentativa até hoje de estudar o nível de conflito de genes individuais. "As pessoas terão duas reações: há mais conflitos do que eu pensava e precisamos aprender a analisá-los melhor", diz Donague, que quer usar o novo método em seu trabalho. No entanto, ele também aponta dificuldades em confirmar a precisão da nova abordagem. Embora a árvore revisada coincida com o que é construído em informações genéticas alternativas, esta última pode revelar suas próprias inconsistências. "Não tenho certeza se sabemos qual é realmente o relacionamento", diz ele. "E se não temos certeza do verdadeiro estado das coisas, não sabemos se conseguimos a árvore certa."
Alterar imagem
Os pesquisadores precisam aplicar a nova técnica mais amplamente para ver como ela pode mudar o conceito de evolução. No entanto, Rokas e Salichos já mostraram que é mais difícil reconstruir galhos curtos da árvore, ou partes "espessas", representando períodos de rápida especiação - especialmente aqueles localizados mais próximos à base da árvore e no fundo da história evolutiva.
"A pesquisa teórica previu esse comportamento, mas nosso estudo pela primeira vez demonstra confirmação usando dados experimentais", disse Rokas.
Rokas argumenta que novas descobertas mudarão a maneira como os pesquisadores interpretam partes pouco claras de uma árvore. “Os biólogos evolucionistas geralmente assumem que, se a árvore não possui os detalhes necessários, ela está errada. E, portanto, se coletarmos mais dados e compormos algoritmos melhores, chegaremos à árvore certa ”, diz ele. Mas a presença de partes conflitantes da árvore que persistem, apesar dos fluxos de dados e da aplicação de um novo tipo de análise, pode indicar a presença de partes espessas. "Acho que em alguns casos o algoritmo será capaz de resolver esse conflito e, em outros, é possível marcar áreas de conflito que dificilmente poderemos resolver."
O estudo dessas partes espessas da árvore pode dar uma nova visão dos estágios especialmente interessantes da evolução, por exemplo, a explosão cambriana, quando a vida passou da predominância de organismos simples para um conjunto variado de espécies animais.
Outros estudiosos concordam que as descobertas podem influenciar a maneira como os especialistas lidam com idéias conflitantes sobre evolução. "Acho que isso é um presságio de uma mudança de paradigma", disse Townsend. "Se usarmos métodos adequados, teremos a oportunidade de aprender mais sobre questões que nos atormentam há muito tempo."
Townsend, que desenvolveu seu próprio método de seleção dos genes mais informativos com base em
sua taxa evolutiva , observa que nem todos os membros da comunidade científica concordam com a necessidade de novas abordagens. "Espero que este trabalho ajude a trazer esta questão para a frente", disse ele.
Escolher a quantidade certa de genes para a construção de protótipos de árvores filogenéticas não é a única questão que atormenta os biólogos da evolução. Eles também precisam concordar com quantas espécies incluir no processamento - quanto mais espécies na árvore, mais difícil será a análise. Os resultados também podem variar devido a diferenças na qualidade dos dados coletados para diferentes espécies. “Se precisamos obter uma verdadeira história evolutiva de como tudo está conectado, então o que é melhor para isso - coletar mais genes ou mais espécies? - diz Donogue. "Eu acho os dois."
Novas abordagens que permitem aos pesquisadores obter resultados precisos usando menos genes podem expandir a árvore evolutiva. A capacidade de selecionar apenas os genes mais informativos pode tornar o processo mais eficiente e permitir que os cientistas criem árvores precisas usando menos dados e recursos. “Se pudéssemos selecionar vários genes e obter a mesma árvore boa que todo o genoma”, diz Khilu, “poderíamos construir uma árvore da vida muito mais detalhada - no nível dos gêneros ou mesmo no nível das espécies - em vez de nos contentarmos com o esqueleto dos ramos mais importantes ".