A análise da afiliação da população humana pelo DNA, em nossa experiência, levanta três grandes questões entre o público: genes e grupos étnicos podem ser interligados, como é a análise da origem de um ponto de vista técnico e se os testes genéticos podem "identificar judeus". Por alguma razão, é precisamente a questão da identidade judaica com o DNA que preocupa tanto aqueles que têm evidências inegáveis de pertencer ao povo escolhido por Deus quanto aqueles que não comem matzo e não leem a Torá.
No novo material da Genotek no Geektimes, tentaremos responder a tudo em ordem. E sim, também definimos judeus.

Raças também conhecidas como grupos populacionais em biologia, medicina e genética
A humanidade tem o mau hábito de justificar a violência com a superioridade "inata" de uma raça sobre outra - é por isso que os biólogos modernos abordam a questão das diferenças genéticas entre as populações com cautela. A (não) existência de fronteiras biológicas entre grupos raciais e étnicos tem sido veementemente discutida ao longo do século XX, mas ainda não foi alcançado um consenso final sobre esse assunto (
1 ).
Esperava-se que a sequência do genoma humano reunisse todos. O genoma, lido de "de" e "para", mostrará que as fronteiras entre os grupos são de natureza social e os genes são os mesmos para todos. O resultado foi diferente: um estudo cuidadoso do código nucleotídico humano reviveu e aumentou o interesse pelas diferenças biológicas entre as populações raciais e étnicas. Os mesmos genes, em geral, encontraram variantes alélicas ligeiramente diferentes associadas ao risco de doenças (
2 ), metabolismo de medicamentos (
3 ), resposta do corpo às condições ambientais (
4 ), e essas variantes foram encontradas em diferentes populações com diferentes frequências.
A busca por genes "indianos" ou "africanos" inexistentes foi interrompida, mas as pesquisas no campo da genética médica e populacional ainda traçam paralelos entre as características biológicas e a etnia dos participantes. O uso dos termos “raça” e “etnia” em tais obras é discutido ativamente (e frequentemente condenado). Houve tentativas de introduzir regras que obrigassem os pesquisadores a justificar a necessidade de usar categorias "escorregadias" e a esclarecer exatamente o que significam termos específicos. Em fevereiro do ano passado, a Science, uma das revistas científicas mais respeitadas, publicou um artigo ambíguo (
5 ), propondo abandonar completamente o uso do termo “raça” na pesquisa genética, substituindo-o por uma “ancestralidade” mais correta e neutra - “origem” .
Mas, mesmo em condições de incerteza com termos, a humanidade ainda é dividida em grupos populacionais: em particular, pela condução correta de ensaios clínicos de medicamentos e pela avaliação do risco de doenças. Por exemplo, três variantes alélicas do gene NOD2 - R702W, G908R e 1007fs - estão associadas a um risco aumentado de doença de Crohn em europeus europeus (
6 ,
7 ), no entanto, nenhuma dessas variantes está associada à doença de Crohn em japonês (
8 ). Sabe-se que os alelos do gene CCR5 afetam a taxa de desenvolvimento da imunodeficiência em pacientes infectados pelo HIV (
9 ): foi encontrada uma opção entre eles que retarda a progressão da doença em americanos de descendência européia, mas acelera seu desenvolvimento em afro-americanos (
10 ). Os asiáticos encontraram uma correlação entre polimorfismos do gene da proteína p53, que regula a resposta ao estresse e suprime o desenvolvimento de tumores, e a temperatura média do inverno nos habitats das populações - adaptação genética à geada (
11 ). E se no passado apenas as informações fornecidas pelos próprios participantes eram usadas para dividir a amostra em grupos étnicos, na era pós-genômica, elas eram cada vez mais suplementadas e refinadas com uma avaliação genética da origem do sujeito.
Variação genética entre populações
Na vida cotidiana, dividimos as pessoas em grupos de acordo com a aparência ou a linguagem da comunicação. A maioria dos dinamarqueses se assemelha mais a cada um deles se assemelha a um italiano (
aqui está uma visualização interessante com retratos médios de diferentes nacionalidades). Os dinamarqueses e italianos estão muito mais próximos um do outro do que cada um deles - dos habitantes da África subsaariana: os fenótipos humanos são agrupados de acordo com o padrão geográfico. A distribuição dos genótipos tem uma estrutura semelhante: os membros de um grupo local, em regra, têm laços familiares mais próximos do que os residentes em áreas remotas, e as populações que vivem em uma região são mais próximas do que aquelas cujos habitats são separados por barreiras geográficas (por exemplo, montanhas ou água array).
Além disso, a diversidade genética da população humana é menor que a de muitas espécies biológicas. Isso se explica pelo fato de a humanidade ser uma espécie jovem: grupos individuais tiveram relativamente pouco tempo para acumular diferenças. Duas pessoas selecionadas aleatoriamente diferem umas das outras por cada um dos ~ 1000 nucleotídeos, enquanto os dois chimpanzés não coincidem uma vez em ~ 500 "letras". E, no entanto, no total, existem cerca de 3 milhões de potenciais "pontos de diferença" no genoma humano. A maioria dessas discrepâncias, chamadas polimorfismos de nucleotídeo único (SNPs)), são neutras ou quase neutras, mas algumas são responsáveis por diferenças fenotípicas entre as pessoas.
A distribuição de polimorfismos neutros (como não possuem significado biológico, não estão sujeitos a seleção evolutiva direcional, são transportados pelo vento das migrações) na população mundial reflete a história demográfica de nossa espécie. Evidências genéticas e arqueológicas indicam que, nos últimos 100.000 anos, o tamanho da população humana cresceu significativamente. As pessoas se estabeleceram fora da África, colonizando o resto do mundo. O processo de reassentamento afetou a distribuição geográfica dos alelos de duas maneiras: primeiro, o "efeito fundador" afetou - na população de imigrantes, em regra, apenas uma parte das variantes genéticas de todo o conjunto de sua diversidade na população ancestral estava representada; em segundo lugar, ocorreu a chamada "travessia sortida", isto é, pares formados principalmente dentro de seu grupo, limitando a distribuição de polimorfismos de novo e emergentes entre indivíduos que habitam diferentes áreas geográficas. Esses processos levaram ao acúmulo gradual de diferenças genéticas.
No contexto de grupos populacionais, os marcadores genômicos começaram a ser estudados nos anos 70 - 80, nos anos 90 começaram a ser usados para identificar a população de uma pessoa em particular. Os pesquisadores demonstraram repetidamente que os polimorfismos genéticos podem isolar com êxito grupos populacionais e determinar a afiliação de um indivíduo. Em seguida, foi demonstrado que as pessoas que vivem no mesmo continente geralmente estão mais próximas geneticamente do que as de diferentes continentes. Inicialmente, nesses estudos, as informações sobre o local de nascimento, raça e etnia eram conhecidas desde o início e eram usadas em conjunto com dados genéticos; se os sujeitos eram distribuídos às cegas entre os grupos apenas com base em características genéticas, a correspondência entre origem geográfica, etnia e estrutura populacional era menos pronunciada. Como estudos posteriores demonstraram, o sucesso dependia dos marcadores genéticos utilizados e do seu número (quanto mais, melhor), da escolha correta das populações de referência e de outros fatores (
12 ).
Em 2004, nos Estados Unidos, a definição genética de população foi usada não apenas em pesquisas biomédicas, mas também em investigações criminais:
este artigo da Nature contém uma história emocionante sobre como policiais, desesperados por encontrar um criminoso, pediram um teste de DNA a uma empresa comercial, decidiram cor da pele do suspeito e abriu o caso. Sugestões para a análise da origem genética atingiram com sucesso a onda de interesse geral das pessoas em seu próprio passado. "Roots mania", assim chamado esse hobby em um artigo da Time, dedicado à "mais recente obsessão da América" - pesquisa genealógica.
Os métodos genômicos são usados ativamente por especialistas que estudam a origem e a evolução dos povos. Por exemplo, em 2013, uma equipe internacional de pesquisadores usou a análise genética para refutar a hipótese da origem dos judeus Ashkenazi dos Khazars (
13 ). O conjunto de dados genômicos usado pelos autores é de domínio público: contém mais de 100 populações mundiais. Propomos simular um pequeno estudo conosco: determinar o local dos clientes da Genotek nesta amostra e, ao mesmo tempo, entender os detalhes técnicos da determinação da população.
Objetivo da pesquisa
Identifique os clientes da Genotek entre as populações de referência. Descubra se há representantes de judeus asquenazes em nossa amostra. Demonstre os princípios e métodos de análise da população de um indivíduo.
Objetivos da pesquisa
Processe os dados de genotipagem de 722 indivíduos usando o programa ADMIXTURE usando o conjunto de dados de Behar et al., 2013 como uma amostra de treinamento.
Materiais e Métodos
O trabalho original de Behar et al., 2013, utilizou dados de 1.774 pessoas: entre eles representantes de 88 populações não-judias (da Arábia, Ásia Central, Extremo Oriente, Europa, Oriente Médio, Norte da África, Sibéria, Sul da Ásia e submarinos). África do Saara) e 18 populações judaicas. Os autores precisavam de um extenso conjunto de dados para determinar com precisão o lugar dos ashkenazes no contexto das populações mundiais: a tarefa era apresentar todas as três regiões geográficas de onde esse grupo poderia vir hipoteticamente - Europa, Oriente Médio e Khazar Khaganate. Os autores enfatizaram a diferença entre a abordagem da amostragem, representando as modernas populações européias, do Oriente Médio e judaicas - descendentes diretos de populações ancestrais e amostras correspondentes ao Khazar Kaganate, que deixou de existir cerca de 1000 anos atrás. O problema é que nenhuma das populações existentes é a herdeira direta do Khaganate. Os autores escolheram residentes do Cáucaso do Sul (abkhazianos, armênios, azerbaijanos, georgianos), do Cáucaso do Norte (Adygs, Bálcares, Chechenos, Cabardins, Ossétios e várias outras nacionalidades), Chuvash e Tártaros como possíveis representantes modernos dos Cazares.
Adicionamos amostras de 722 pessoas de várias regiões da Rússia ao conjunto de dados.
Para análise estatística, foi utilizado o programa ADMIXTURE, que permite estimar a origem mais provável de um indivíduo com base nos dados dos genótipos. Além disso, os autores do artigo em discussão usaram outros métodos estatísticos que deram uma resposta semelhante à questão colocada. Vamos nos concentrar no ADMIXTURE, pois é esse algoritmo que nos permite estimar a contribuição percentual das populações ancestrais para os genomas estudados.
ADMIXTURE usa métodos de Monte Carlo em cadeias de Markov (cadeia de Markov Monte Carlo, MCMC). Aqui está um
link para um artigo dos autores do algoritmo para quem deseja entender com mais detalhes o lado matemático do processo.
Vamos ver como o ADMIXTURE funciona no exemplo de amostras e populações do nosso conjunto
No total, temos 2.496 amostras / indivíduos, cada um pertencendo a uma das 106 populações modernas. Sugerimos que as populações modernas provavelmente descendem de um número relativamente pequeno de populações ancestrais. As “populações ancestrais” nesta análise são alguns aglomerados genômicos antigos, unidos pelo princípio da similaridade genética. O ADMIXTURE permite apresentar arbitrariamente suposições sobre o número de tais clusters na amostra e selecionar o número ideal que melhor descreve a distribuição real dos dados genômicos.
Tendo recebido informações sobre os genótipos e o número estimado de populações “ancestrais” (K), o ADMIXTURE constrói um modelo que estima a contribuição de cada uma das populações “ancestrais” para cada amostra. Ao interpretar os dados, tanto a composição quantitativa do genoma (porcentagem de aglomerados) quanto a qualitativa são importantes - sua presença ou ausência em genomas específicos. Com base nesses dados, é possível fazer suposições sobre processos evolutivos em uma população, em particular sobre a presença ou ausência de “raízes” comuns em grupos populacionais. No entanto, as conclusões serão legítimas se o modelo que construímos for bom: o valor ótimo de K. é selecionado.
Selecionamos o valor ideal de K
Como determinar quantas populações "ancestrais" se aproximam mais da verdade de uma determinada amostra? Empiricamente!
ADMIXTURE é um programa inteligente: construindo um modelo da estrutura genética de populações com base em dados sobre os genótipos de indivíduos (avaliando a contribuição de cada um dos agrupamentos genômicos antigos para cada um dos genomas da amostra) para um determinado número de K, ela não esquece de fazer uma comparação com a realidade no final. Verifique quão bem a entrada é descrita pelo modelo construído. Uma medida de comparação é o "erro" - um valor que descreve a incompatibilidade entre o modelo e os dados reais. Quanto maior o erro, pior é a suposição do número de populações ancestrais.
Como escolher o valor ideal de K? Iniciamos o algoritmo ADMIXTURE nesta amostra, substituindo valores diferentes de K, e obtemos para cada K seu próprio valor de erro. Traçamos a dependência da magnitude do erro em K. Aqui está o gráfico obtido pelos autores do artigo:
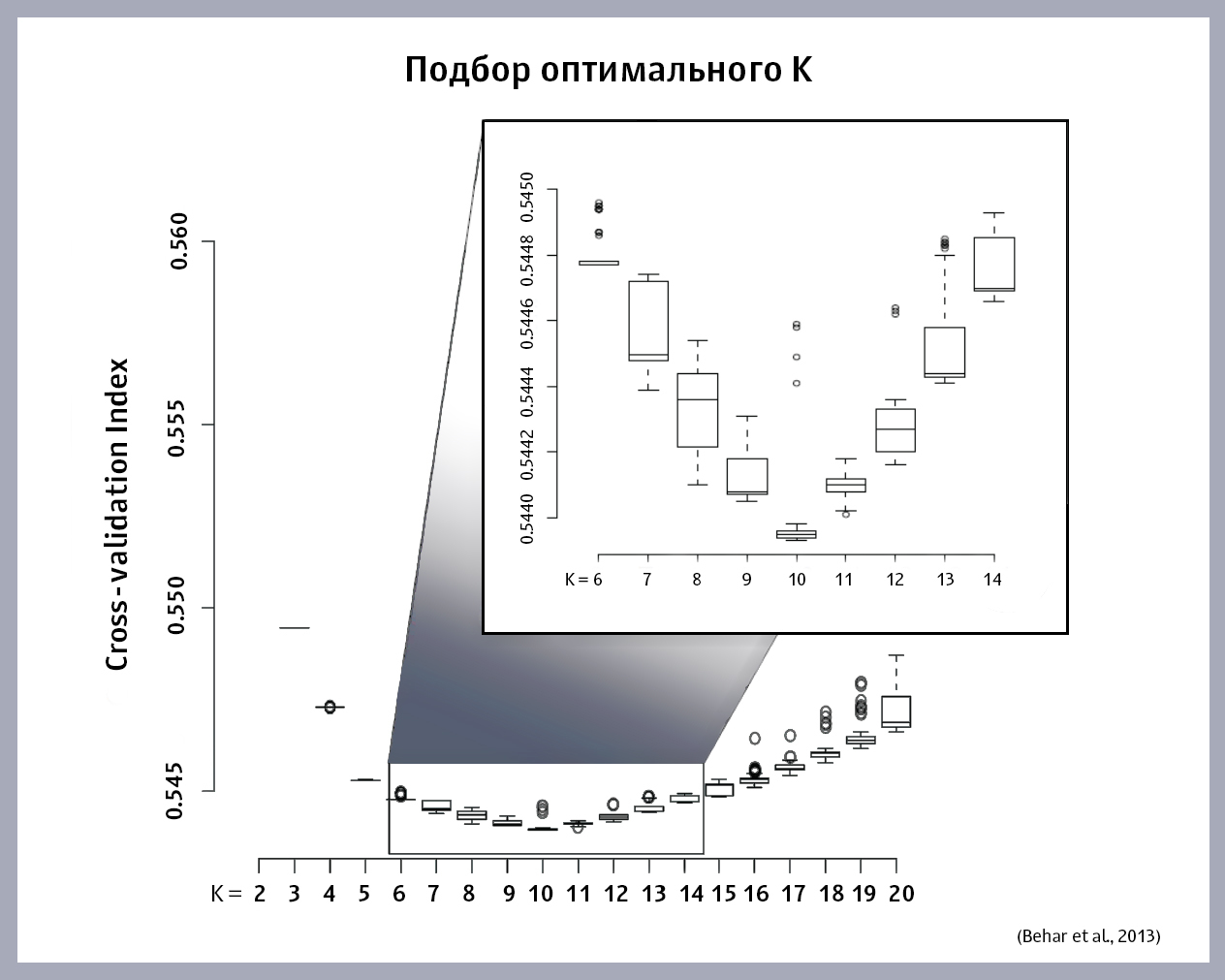
O valor ideal de K está no ponto mínimo da função. Se o mínimo não for encontrado no gráfico (a função está constantemente aumentando ou diminuindo), você terá que construir modelos escolhendo novos Ks até encontrar o correto.
Mesmo com o K selecionado de maneira ideal, a confiabilidade dos resultados da análise depende da exatidão da amostra:
1. Os indivíduos não devem estar relacionados um com o outro.
2. Os polimorfismos de nucleotídeo único (SNPs) usados para genotipagem devem ser distribuídos uniformemente sobre o genoma com uma densidade suficientemente alta.
3. Os alelos SNP devem estar em ligação de equilíbrio, ou seja, a probabilidade da presença de um dado alelo em um indivíduo em particular deve depender apenas da frequência desse alelo na população e não de outros alelos no genoma.
Como pode ser visto no gráfico, o K ideal para esta amostra foram 10 populações "ancestrais".
Resultados
Os resultados da análise são visualizados pelo ADMIXTURE assim (na figura apenas uma parte dos dados é visível):
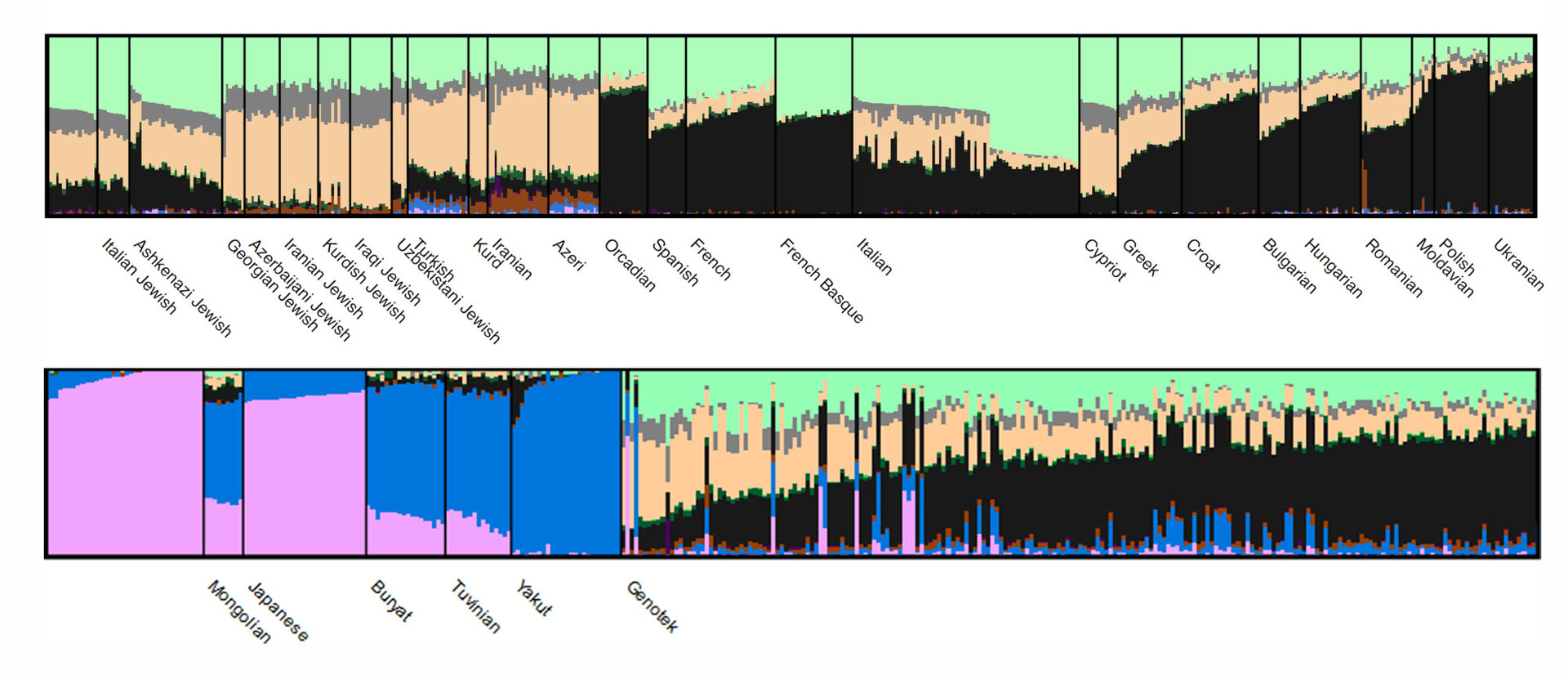
Cada cluster tem sua própria cor e as populações diferem (ou não) nas ações dos clusters no genoma.
Aqui está a versão interativa da imagem para um estudo detalhado: mova o mouse e role para ver todas as populações ou considerar alguns dos grupos em mais detalhes.
Em geral, dentro da “população” de Genotek, espera-se que a proporção de agrupamentos corresponda ao padrão característico de populações de origem européia oriental. O interessante começa no nível das amostras individuais:
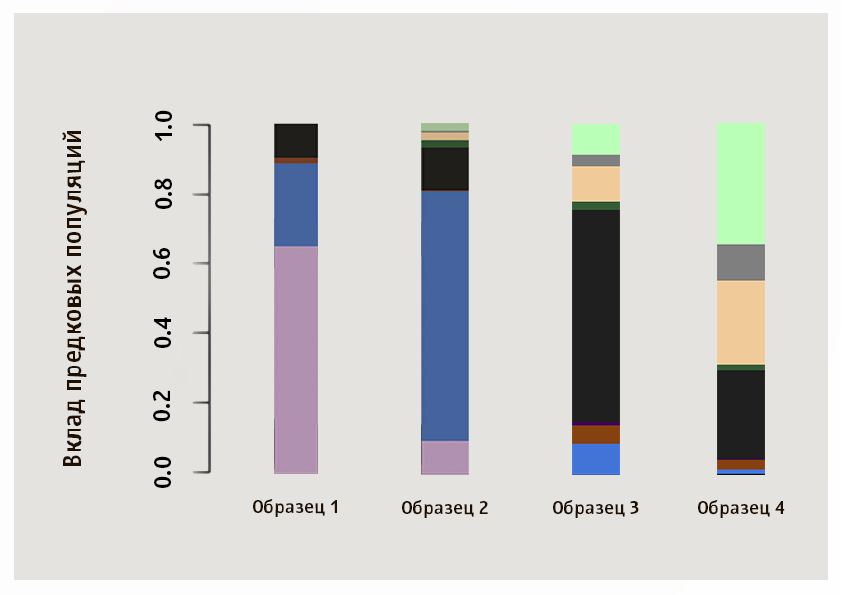
Embora a população mais próxima da amostra seja determinada por valores numéricos, muitas informações também podem ser obtidas por comparação visual de padrões. Sugerimos que você determine independentemente as populações mais próximas para amostras de quatro clientes Genotek a partir da imagem.
A respostaNesta figura, as amostras 1 e 2 são de origem asiática: a predominância do aglomerado rosa é típica para os japoneses e os Khan em nossa amostra, azul para os Yakuts, a terceira amostra mostra a proporção de componentes típicos de russos, bielorrussos, ucranianos e poloneses, e a quarta é típica Judeu Ashkenaz.
Das 722 amostras, encontramos 9 judeus Ashkenazi.
Conclusão
A afiliação populacional está longe de ser o único fator que determina a auto-identificação étnica de uma pessoa. No entanto, ainda é possível revelar uma correlação entre grupos étnicos e a estrutura do genoma de seus representantes. Essa análise é usada para propósitos científicos e médicos e para o estudo de suas próprias raízes por todos os que chegam. Ao mesmo tempo, é importante entender que os modelos estão sendo aprimorados constantemente, e os resultados obtidos para maior precisão devem ser considerados em conjunto com outros dados, por exemplo, a árvore genealógica da família.
Não foram encontradas evidências da origem Khazar de Ashkenazi pelos autores do artigo original. Os testes genéticos, é claro, “sabem” como identificar judeus - no entanto, não se deve esquecer que o “judaísmo” é, antes de tudo, um estado de espírito.
Num futuro próximo, o teste atualizado do DNA de genealogia, com resultados estendidos, será lançado na Genotek: elevaremos o número de populações para centenas, acrescentando populações judaicas. Atualizaremos as informações em sua conta pessoal para todos que já passaram seu material genético. Se você ainda não está genotipado, convidamos você a
participar .
Referências
- Foster M., Sharp R. (2002). Raça, Etnia e Genômica: Classificações Sociais como Proxies da Heterogeneidade Biológica. Genome Res.
- Collins FS, McKusick VA (2001). Implicações do Projeto Genoma Humano para a ciência médica. JAMA.
- Nebert DW, Menon AG (2001) Farmacogenômica, etnia e genes de susceptibilidade. Farmacogenômica J.
- Olden K., Guthrie J. (2001). Genômica: implicações para a toxicologia. Mutat. Res.
- Yudell M., Roberts D., DeSalle R., Tishkoff S. (2016). Tirando a raça da genética humana. Ciência.
- Ogura, Y. et ai. (2001) Uma mutação de mudança de quadro no NOD2 associada à suscetibilidade à doença de Crohn. Natureza.
- Hugot, JP et ai. (2001) Associação de variantes de repetição ricas em leucina NOD2 com suscetibilidade à doença de Crohn. Natureza.
- Inoue, N. (2002). Falta de variantes comuns de NOD2 em pacientes japoneses com doença de Crohn. Gastroenterologia.
- Martin, MP e outros (1998). Aceleração genética da progressão da AIDS por uma variante promotora do CCR5. Ciência.
- Gonzalez, E. et al. (1999). Efeitos modificadores da doença por HIV-1, específicos da raça, associados aos haplótipos do CCR5. Proc. Natl Acad. Sci. EUA
- Shi, Hong et al. (2009). A temperatura do inverno e os raios UV estão intimamente ligados a alterações genéticas na via do supressor de tumores p53 no leste da Ásia. American Journal of Human Genetics.
- Bamshad M., Wooding S., Salisbury B. et al. (2004). Desconstruindo a relação entre genética e raça. Nat Rev Genet.
- Behar DM et al. (2013). Nenhuma evidência de dados genômicos de uma origem Khazar para os judeus asquenazes. Biologia Humana