 Você pode dizer o quanto quiser que há algo errado com a medicina na Rússia, mas nossa proteção ao consumidor é uma das melhores do mundo.
Você pode dizer o quanto quiser que há algo errado com a medicina na Rússia, mas nossa proteção ao consumidor é uma das melhores do mundo. Comparado aos nossos padrões europeus para dispositivos médicos, é apenas um problema.
Nos últimos anos, temos que provar qualquer efeito. Ou seja, se você se deparar com uma pomada de rugas, mas ela não remover rugas, poderá reclamar com o Rospotrebnadzor. E em algum lugar distante da indústria, você pode ouvir o som de uma jarra de vaselina sendo aberta. Se você se sentir doente depois de tomar um bálsamo cicatrizante, também poderá reclamar. E novamente - um povo, eles vão chegar a alguém com um cheque.
Toda essa história de requisitos de resistência nos afetou seriamente alguns anos atrás, quando registramos o Blefarogel. Mas deixe-me apenas contar em detalhes a quantidade de vaselina, lágrimas e ranho que gastamos nesse processo.
4 círculos do inferno
A base para o registro de dispositivos médicos é o
Decreto nº 1416 .
Ele descreve as regras e procedimentos para o registro. Para passar por esse procedimento bastante complicado, os seguintes testes precisam ser realizados:
- Toxicológico (comprovando que o produto é seguro em termos de materiais e substâncias utilizados na fórmula).
- Técnico (comprovando que o produto, em suas propriedades e características técnicas, atende aos requisitos da TU e da documentação operacional do fabricante, além do fato de que o processo de produção será uniforme e que não será necessário verificar cada elemento individual do lote).
- Clínico (comprovando a eficácia e segurança da solução de um problema médico).
- E um exame de qualidade, eficiência e segurança, onde a comissão coleta todos os dados do produto de uma só vez e decide sobre a possibilidade de seu registro e liberação no mercado.
Um ponto importante:
por lei, um dispositivo médico não pode curar. Ou seja, se tiver um efeito terapêutico, este medicamento é uma categoria completamente diferente. O dispositivo médico pode ser algo auxiliar no processo. Por exemplo, se um tonômetro for registrado, deve-se provar que mede a pressão, que a manga nos pontos de contato com a pele não causará alergias ou várias dermatites, que a pressão será medida com uma determinada tolerância durante toda a vida útil do dispositivo. No caso de um pato, é necessário provar sua hipoalergenicidade e segurança para o paciente. No caso dos nossos géis, o principal é passar por Roszdravnadzor, incluindo a toxicologia.
A toxicologia costumava ser relativamente simples. Hoje, precisamos começar com a análise de componentes e suas interações. O que é importante, isso diz respeito não apenas ao produto em si, mas também à embalagem. Anteriormente, eles apenas verificavam o gel e, agora, de acordo com os novos padrões, o gel e a embalagem (garrafa com tampa) de todas as embalagens e cores usadas, bem como a interação. Isso é feito para que a substância do frasco não altere as propriedades do gel, migrando para sua composição.
Primeiro, é necessário realizar uma revisão da literatura, ou seja, encontrar informações na forma de artigos e análises sobre análogos registrados que provam sua eficácia e segurança, encontrar práticas sobre substâncias ativas e provar que elas são seguras na mistura.
Por que a primeira etapa da revisão literária? Deixe-me lembrá-lo de que não temos um medicamento, mas um dispositivo médico. Não há necessidade de testar a nova fórmula. Mais frequentemente do que não.
É assim: nos referimos a artigos para exame, ou seja, encontramos revistas nos arquivos, notificamos capturas de tela e cópias para que a própria comissão não pareça mais tarde.
Então, precisamos de feedback da prática clínica sobre análogos registrados. Ou seja, precisamos encontrar publicações (ou pegar o analógico mais próximo já registrado uma vez e verificar como ele resolve esse problema). Com base nas informações coletadas sobre análogos registrados, é elaborado um ato de avaliação dos resultados de ensaios clínicos de dispositivos médicos.
Em seguida, com um pacote de documentos confirmando a conclusão de todas as etapas indicadas acima, além de outros documentos adicionais para o dispositivo médico, somos enviados a Roszdravnadzor para registro.
Como está indo o processo?
Primeiro, um pedido e um dossiê para o produto futuro são submetidos à Roszdravnadzor. O aplicativo descreve o objetivo, classe de risco, escopo e outras informações sobre o dispositivo médico, além de informações sobre o fabricante. Além do aplicativo, é anexado um conjunto de documentos: protocolos, certificados de todos os testes realizados, documentação técnica (TU), documentação operacional (instruções de uso, etiquetas), imagens fotográficas, um relatório de análise de risco, informações sobre documentos regulamentares, um certificado de teste de qualificação e contratos de locação e outros documentos do fabricante.
Ou seja, no momento da submissão do pedido, é necessário concluir todos os testes internos (em nossa produção, em nosso próprio laboratório, eles são mais difíceis do que em laboratórios estaduais, porque é fundamental mais lucrativo ir na primeira vez e não encontrar revisões de partes posteriormente no processo de vendas). Em seguida, passe a toxicologia para um dos laboratórios com acreditação estadual.
No nosso caso, por exemplo, o
Blefarogel (um meio de higiene das pálpebras na prevenção
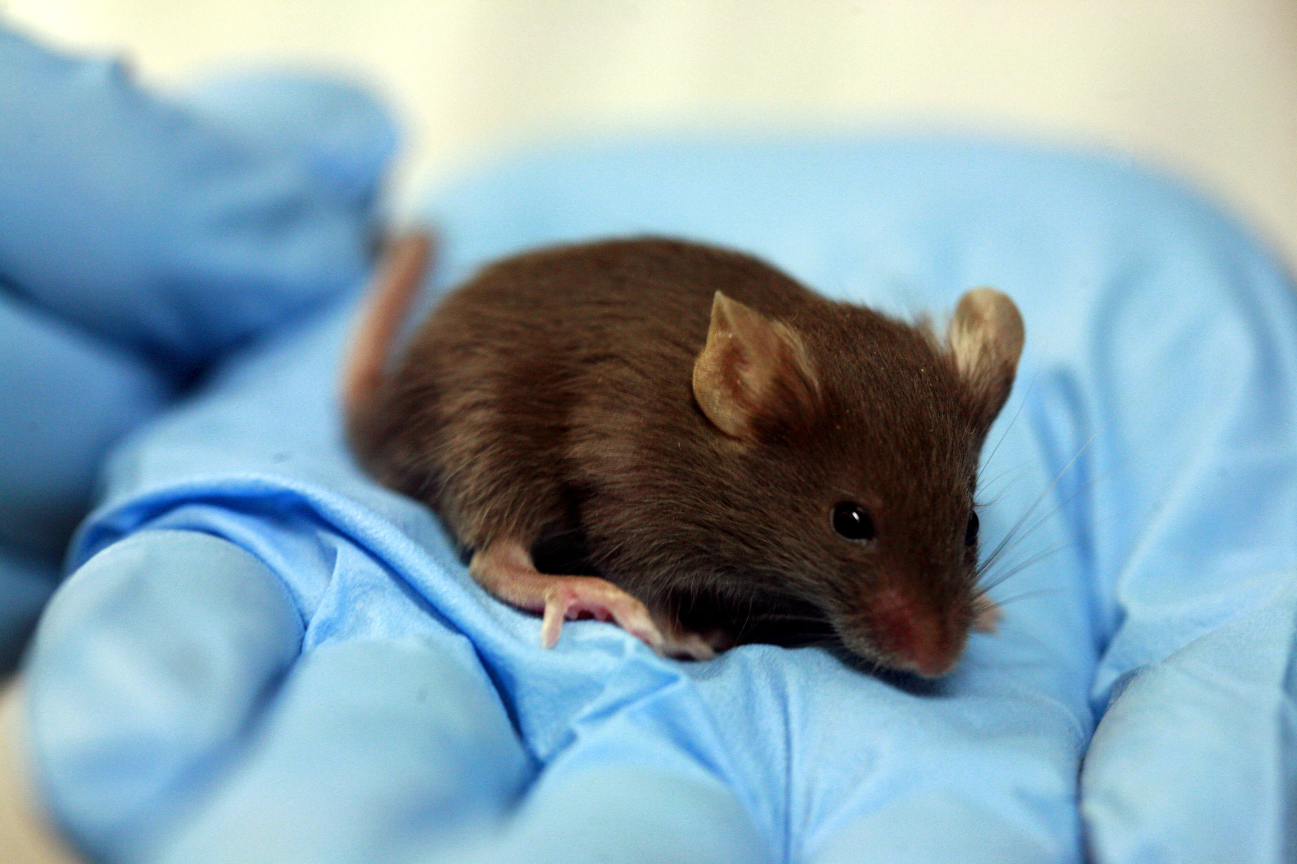
da síndrome do olho seco ) é um gel que é aplicado nas pálpebras e na pele do rosto. Toxicologia eram camundongos. Não sei exatamente o que eles fizeram com eles, mas suponho que eles rasparam duas áreas de cada animal, uma manchada de gel, a segunda esquerda ou manchada de algo neutro. E assistiu o mouse. Felizmente, eles não empurraram dentro do mouse. (Ok, eles quase não o empurravam - o fato é que na ficha de dados de segurança [ou MSDS - ficha de dados de segurança do material]) de propileno glicol há dados sobre a dose letal quando tomados por via oral. Ele precisa de 4 gramas por mouse [ele quebra fisicamente ao mesmo tempo provavelmente] E, afinal, de alguma forma, eles definem essa dose ...) Mas voltemos aos produtos de mouse mais bem-sucedidos, que são simplesmente borrados. Eles têm vizinhos que receberam garrafas limpas do produto, nas quais despejam água e se defendem. Em seguida, elas são borradas com a água das garrafas para verificar se o plástico não libera algo perigoso ao longo do tempo. Ou não: a metodologia exata não foi divulgada aos fabricantes, existe uma alta probabilidade de que essa água da garrafa seja simplesmente analisada quimicamente e, mesmo se houver suspeita, seja injetada por via intradérmica.
Sentimos muito pelos ratos, mas a abordagem é a seguinte: se não fosse pelos nossos testes, eles não teriam nascido. Em geral, eles não são atormentados por lá (pelo menos, já entramos com um estudo pronto sobre nós mesmos). Também sabemos que os assistentes de laboratório têm sentimentos calorosos por ratos, comparáveis ao amor de um aldeão por frango. E suspeitamos que, ao final da pesquisa, o mouse não esteja completamente descartado, mas seja enviado para reabilitação antes de novos testes. Mas isso é apenas uma teoria: se houver alguém aqui que possa contar sobre seu destino, seria maravilhoso.
A mesma aplicação deve ser acompanhada de pesquisa técnica. Especificamente, escreva TU (documento regulamentar para um dispositivo médico que estabelece requisitos técnicos) e comprove que o dispositivo médico corresponde a ele. Os especialistas concordam com nossas condições. Em seguida, a estabilidade das amostras seriais e a conformidade de seus dossiês de produtos são verificadas: cor, cheiro, condutividade elétrica, atenuação do sinal ultrassônico e assim por diante - tudo isso é indicado. Nesse estágio, geralmente é necessário reformular uma palavra específica na descrição do produto. Nosso Mediagel tem uma viscosidade diferente: este é um certificado de registro, mas três conjuntos de teste são para viscosidade média, gel mais líquido e mais denso.
Realizamos as mesmas verificações regulares quanto à conformidade com a TU em nossa própria produção. Somente uma vez em toda a prática houve um caso em que nos aproximamos dos limites permitidos do teste, geralmente andamos exatamente no centro das tolerâncias entre os limites.
A classe de risco do produto é determinada por sua finalidade, método de uso, duração do uso, invasividade e outros critérios. Temos géis de ultrassom, por exemplo, a duração do contato com a pele até uma hora é a primeira classe. E na segunda classe, por exemplo, espelhos ginecológicos, coisas diferentes para os recém-nascidos caem.
Há uma diferença nos ensaios clínicos. Primeira classe - ensaios clínicos podem ser realizados imediatamente. A segunda classe e acima - a clínica somente após a permissão oficial de Roszdravnadzor. Antes dos ensaios clínicos, a equipe de desenvolvimento tem o direito de testar apenas em si mesmos. O objetivo é verificar o efeito principal. O objetivo da versão beta é entender os possíveis efeitos colaterais e vários casos interessantes, como "enquanto estiver tomando outros medicamentos". Os ensaios clínicos começam quando a segurança do dispositivo médico é comprovada em teoria.
Observo que, se não se trata de produtos médicos, mas de cosméticos, são realizados estudos com voluntários em clínicas credenciadas para pesquisas científicas desse tipo.
Na prática, é o seguinte: a comissão conclui a análise dos documentos (com exceção dos dispositivos médicos da 1ª classe de risco e dispositivos médicos para diagnóstico in vitro); depois, eles enviam uma carta e dão 50 dias úteis para realizar estudos clínicos e fornecer os resultados. Para a maioria dos nossos géis, toda a prática clínica foi obtida mesmo antes do aperto dos requisitos, portanto não houve dificuldades. Mas agora temos uma ordem alvo dos oftalmologistas para o novo Blefarogel Forte - é como o Blefarogel-2 com compostos de enxofre para o demodex, apenas para casos avançados. Se o Blefarogel-1 for “para que meus olhos não doam enquanto eu estou sentado no computador”, o Blefarogel-2 é “para que não haja inflamação do carrapato e ele não tenha nada para comer para sempre”, o Blefarogel-Forte será “genocídio do carrapato a todo custo”. No sentido em que aumentamos deliberadamente o risco de alergias, aumentando a proporção de substâncias ativas, mas recombinamos a fórmula para que ela ajude em casos especialmente difíceis. Como isso é feito a pedido de médicos, que apenas antecipam que precisam dessa ferramenta.
Fim do épico
Em seguida, a integridade dos documentos é verificada em Roszdravnadzor: um consultor executivo que simplesmente verifica o pacote se destaca. Os documentos são digitalizados e enviados para uma organização especializada no departamento de Roszdravnadzor. Em consequência, é estabelecida uma conclusão em Roszdrav com base na totalidade de todos os documentos. Torna um grupo de especialistas. Se tudo estiver bem e os documentos corresponderem um ao outro, eles dão permissão para o registro do estado.
A partir deste momento, você não pode tocar em nada. Substitua a letra na etiqueta ou substitua o ícone automaticamente significa que você precisa se registrar novamente. Nos pediram para alterar de alguma forma o rótulo de Mediagel-s para russo-inglês. Isso significa se registrar novamente. Seis meses foram necessários para inserir o texto em inglês.
Como o processo é especialista, está amplamente associado a interpretações específicas de normas bastante complexas. Isso significa que várias vezes você pode enviar documentos de acordo com o mesmo esquema e, em seguida, é necessário um novo pedaço de papel. Em geral, esse é um tipo de acidente: eles podem ser encerrados a cada reenvio. Ao mesmo tempo, o antigo registro estadual permanece e não desaparece, mas perdemos dinheiro para julgamentos e pagamos um grande imposto estadual mesmo para registro e alterações. E nós gastamos ratos do estado.
Em seguida, você precisa aguardar o pedido no papel. Quando houver um pedido, o produto irá para o registro, ou seja, para o
site em que as farmácias analisam o que é o quê. Somente a partir deste momento você pode fazer algo. A farmácia não aceita produtos que não estão na EF do Ministério da Saúde, se não forem cosméticos, é claro.
Somente nesse momento a produção pode começar. Sem o número do Ministério da Saúde, não podemos apenas vender, mas até produzir na Rússia.
Tudo, a partir deste momento, estamos fazendo o dispositivo médico em série. Por experiência: é muito importante ter um laboratório de controle de qualidade bem desenvolvido, porque qualquer reclamação significa uma verificação completa na fábrica (a julgar pelo comportamento, você não pode retornar sem um relatório de pelo menos algumas violações) e interromper os embarques. Nesse sentido, preferimos realizar os testes de aceitação e testes periódicos por conta própria e encerrar os estudos com pequenas amostras. Quero dizer que 10 voluntários do padrão são poucos, mas 100 voluntários já são uma amostra mais ou menos representativa. A longo prazo, uma pesquisa mais extensa sempre compensa, e para mim é um grande mistério o motivo pelo qual outros fabricantes nem sempre fazem isso.
No próximo post, tentarei falar sobre outra etapa importante da pesquisa - o teste de desafio: é quando as amostras são especialmente contaminadas por bactérias patogênicas. Essa parte da pesquisa nunca deve ser realizada em qualquer lugar próximo aos laboratórios de produção ou pesquisa, para que os testes de desafio sejam realizados.