Mitocôndrias - pequenos trabalhadores ou grandes chefes?Se você acha que a história mais importante para vivermos juntos começa durante o casamento, esse não é o caso. A história mais importante da vida de cada pessoa começou há mais de um bilhão de anos atrás, quando nossos ancestrais unicelulares distantes foram forçados a assinar um "contrato de casamento" com aqueles a quem chamamos agora de mitocôndrias (veja a teoria da simbiogênese).
As mitocôndrias têm duas membranas (internas e externas) e seu próprio material hereditário na forma de DNA (Fig. 1). Na membrana interna das mitocôndrias existe um sistema de fosforilação oxidativa, cuja operação fornece a oxidação de substratos energéticos com a formação de ATP.
Fig. 1 Estrutura esquemática das mitocôndrias
No contrato de casamento da célula e das mitocôndrias, não há cláusula "em doença e saúde" - e é bom. Se a mitocôndria envelhece, a célula pode matá-la durante a mitofagia, e as mitocôndrias, por sua vez, regulam o processo de apoptose nas células disfuncionais e antigas. Se o processo de controle mútuo da qualidade for interrompido, serão lançados mecanismos de envelhecimento. Os mecanismos de apoptose são interrompidos, aumenta o número de radicais livres não controlados pelas mitocôndrias. Isso causa inflamação sistêmica, danos ao DNA da célula. Assim, existe uma forte relação entre disfunção MX, doenças relacionadas à idade, envelhecimento e disfunções metabólicas [1]. A disfunção metabólica é um constante condutor do apocalipse do envelhecimento.
“Como um esquilo em uma roda” - a dinâmica das mitocôndrias
Nem toda a culpa por distúrbios metabólicos recai sobre nossos excessos. Os distúrbios metabólicos estão associados principalmente à incapacidade das mitocôndrias de lidar com os nutrientes. As mitocôndrias na célula não são fáceis. Nós "alimentamos" nossas células em excesso ou muito pouco e apresentamos a elas uma "solicitação" para fornecer energia na forma de ATP, cuja quantidade deve corresponder exatamente às nossas necessidades. Para "sair" regularmente dessa situação, as mitocôndrias realmente usam alguns "movimentos" - fissão e fusão. Essas “mitodomoções” são unidas sob o nome “dinâmica mitocondrial”. O equilíbrio entre divisão mitocondrial e fusão é o mecanismo central de adaptação bioenergética às necessidades metabólicas da célula [2, 3].
A maioria das mitocôndrias é encontrada em tecidos com alta necessidade de energia - músculos, fígado, tecido adiposo marrom e cérebro. Não é de surpreender que a dinâmica das mitocôndrias nesses tecidos tenha sido melhor estudada.
Portanto, se uma célula de qualquer um desses tecidos (exceto alguns neurônios no cérebro, mais sobre isso mais tarde) recebe uma grande quantidade de nutrientes (a ingestão excede o custo), as mitocôndrias estão em um estado dividido (fragmentado). Se a célula está em um estado de fome (renda menor que o custo), as mitocôndrias se fundem e elas estão em um estado conectado. [3,4] É assim que a homeostase celular é mantida (fig. 2).
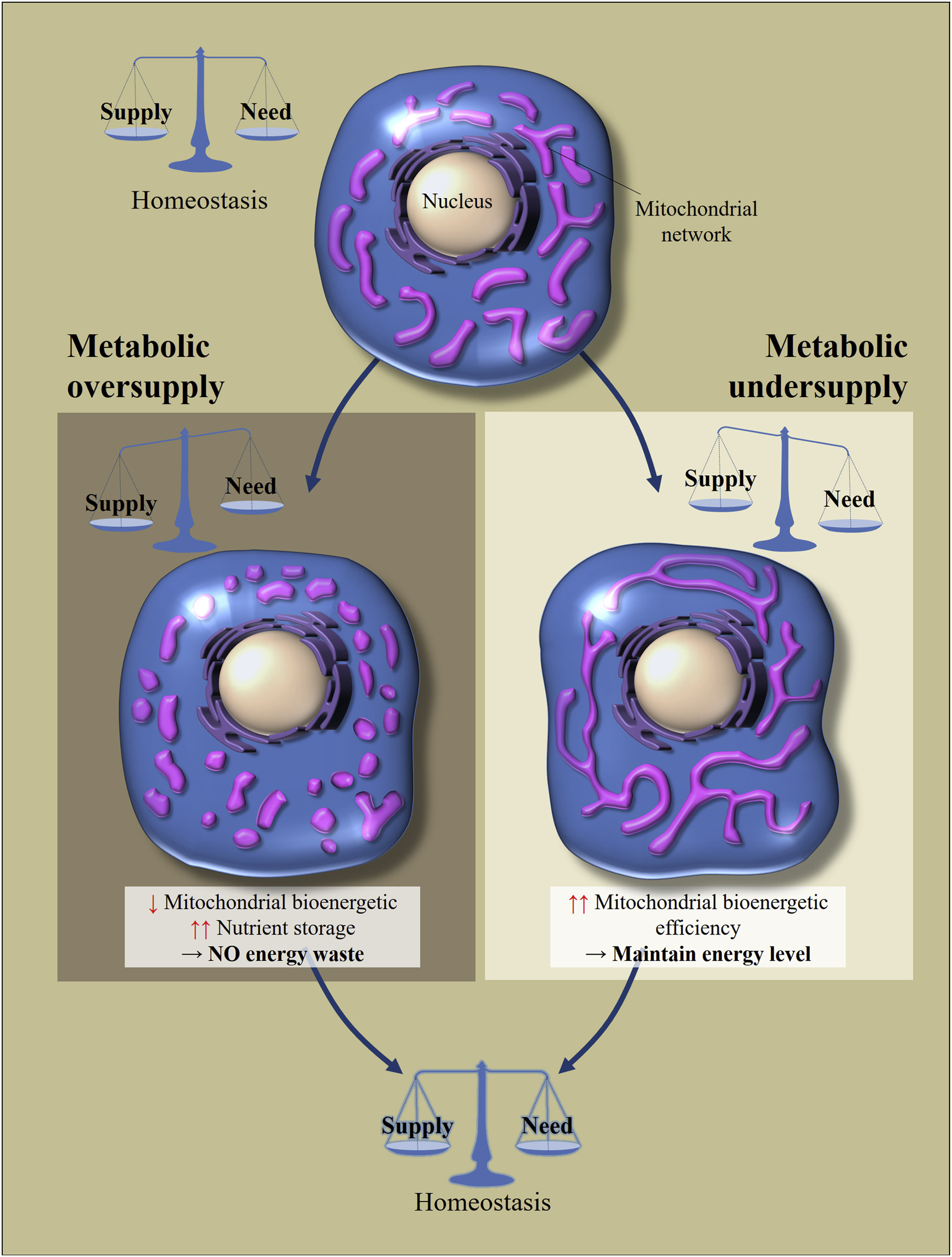 Fig. 2
Fig. 2 Regulação da morfologia e eficiência da bioenergia das mitocôndrias em resposta ao consumo excessivo ou insuficiente de nutrientes [em 2]
A homeostase metabólica celular depende do equilíbrio entre a ingestão de nutrientes e seu consumo. Alterações no suprimento de nutrientes levam a adaptações celulares para restaurar o equilíbrio. O excesso de nutrição leva à fragmentação da rede mitocondrial, o que causa uma diminuição na eficiência bioenergética das mitocôndrias. Isso evitará a perda de energia. Por outro lado, com a fome metabólica, as mitocôndrias ficam mais longas para aumentar sua eficiência bioenergética.Qual é o truque desses movimentos? Se a célula estiver com fome, a fusão das mitocôndrias pode aumentar sua eficiência em bioenergia (a quantidade de ATP criada por molécula de nutriente). Se um excesso de nutrientes entra na célula, eles podem ser 1) armazenados ou 2) dissipar essa energia na forma de calor. A tarefa das mitocôndrias nesse caso é dissipar mais energia na forma de calor, armazenar menos na forma de ATP (o acúmulo de NADH e ROS levará ao estresse oxidativo). A fragmentação das mitocôndrias lhes permite reduzir a eficiência da bioenergia, cujo principal mecanismo de redução é considerado "vazamento" de prótons.
Então, vamos trabalhar, e a vida das mitocôndrias prossegue constantemente no ciclo de divisão e fusão (Fig. 3).
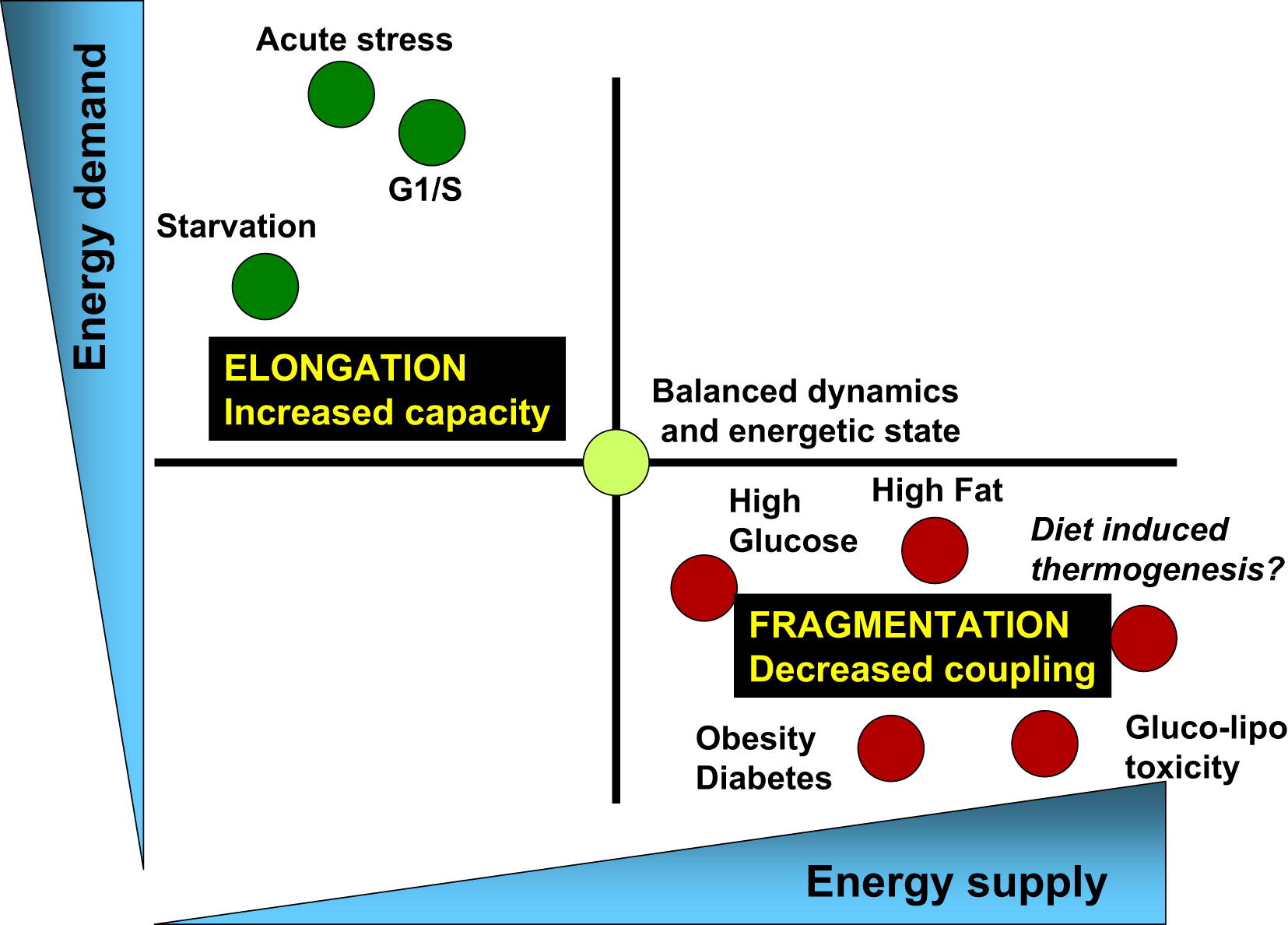 Fig. 3 O equilíbrio do consumo de energia e do suprimento de energia está associado a alterações correspondentes na arquitetura das mitocôndrias e sua eficiência em bioenergia [de 3]
Fig. 3 O equilíbrio do consumo de energia e do suprimento de energia está associado a alterações correspondentes na arquitetura das mitocôndrias e sua eficiência em bioenergia [de 3]
Os processos fisiológicos associados a um aumento na demanda de energia e uma diminuição no suprimento de energia (por exemplo, estresse agudo, fome e fase G1 / S) são caracterizados pelo alongamento das mitocôndrias e respiração associadas à síntese de ATP. Por outro lado, processos fisiológicos associados à diminuição da demanda de energia e ao aumento da oferta (altos níveis de nutrientes, obesidade e diabetes tipo 2) estão associados à fragmentação das mitocôndrias, produção de calor ou diminuição da função mitocondrial.Ciclos saudáveis de fissão e fusão são essenciais para a saúde metabólica celular
O ciclo normal de divisão e fusão mitocondrial é um elemento-chave em seu controle de qualidade. Porque Durante a divisão mitocondrial, duas filhas são formadas, uma das quais tem maior potencial de membrana e vai além no ciclo de divisão de fusão, e a outra, com uma membrana mais despolarizada, permanece separada até que o potencial de membrana seja restaurado. Se o potencial é restaurado, ele se reúne com a rede mitocondrial. Se permanecer despolarizado, é eliminado no processo de autofagia, que é a chave para a qualidade do pool mitocondrial (Fig. 4).
A inibição a longo prazo da divisão mitocondrial (com fome prolongada de células) leva ao acúmulo de mitocôndrias danificadas, que não podem ser segregadas [3, 4].
Por outro lado, um excesso de nutrientes leva à inibição da fusão mitocondrial, o que leva à interrupção do ciclo da dinâmica mitocondrial, aumenta a heterogeneidade mitocondrial intracelular. Sim, com excesso de alimentos, a fragmentação mitocondrial é protetora, mas a fragmentação prolongada, como a fusão prolongada, é prejudicial ao controle de qualidade mitocondrial. Não há remoção seletiva, a massa mitocondrial diminuirá e consistirá em pequenas mitocôndrias despolarizadas.
 Fig. 4 Ciclo de vida das mitocôndrias e sua regulação da disponibilidade de nutrientes [de 3]
Fig. 4 Ciclo de vida das mitocôndrias e sua regulação da disponibilidade de nutrientes [de 3]As mitofusinas não são apenas proteínas
No nível molecular, a fusão mitocondrial é um processo de dois estágios que requer uma fusão coordenada das membranas externa e interna durante eventos seqüenciais separados. Nos mamíferos, esse processo é regulado por três proteínas pertencentes às GTPases: Mfn1 e Mfn2 são necessários para a fusão da membrana externa e OPA1 - para a fusão da membrana interna. Outras proteínas são necessárias para a divisão, Fis1 e Drp1.
O papel das proteínas mitofusinas foi estudado em estudos de perda e ganho de função. Os ratos mutantes das proteínas mitofusinas morrem logo no meio da gestação, porque a fusão mitocondrial se torna impossível para elas. As mitofusinas são importantes para os processos de autofagia e mitofagia. A expressão diminuída de Mfn2 nos cardiomiócitos bloqueia o início do processo de autofagia, porque a fusão dos autofagossomos com os lisossomos está bloqueada. A depleção de Mfn2 leva a uma diminuição no potencial das membranas mitocondriais; para compensar, há uma diminuição na cadeia respiratória, a captação de glicose aumenta e a síntese de glicogênio diminui. A célula muda para a glicocólise anaeróbica, e esse é o caminho para a degeneração oncológica da célula. A deficiência de Mfn2 leva a alterações neurodegenerativas. Um aumento na expressão de Mfn2 nos músculos esqueléticos aumenta sua sensibilidade à insulina.
Mfn1 desempenha funções semelhantes, mas provavelmente em outros tecidos (a expressão de Mfn2 e Mfn1 varia em tecidos diferentes) - Mfn1 é expresso mais no coração, fígado, pâncreas, testículos e Mfn2 no coração, músculo esquelético, cérebro, tecido adiposo marrom .
Assim, as mitofusinas são reguladores chave da dinâmica mitocondrial. A expressão das mitofusinas é diferente em diferentes órgãos, elas fornecem eficiência em bioenergia e mecanismos de adaptação à disponibilidade de nutrientes, e o "destino" da célula depende deles. Não é de surpreender que as proteínas de fusão mitocondrial sejam alvos potenciais para intervenções farmacológicas [2, 5].
Hipotálamo, mitocôndrias, disfunção metabólica e envelhecimento
A dinâmica das mitocôndrias é importante em todas as células. Nas células beta do pâncreas, as mitocôndrias são sensores de nutrientes e geradores de sinais de síntese de insulina; nos músculos, a dinâmica mitocondrial é importante para a regulação do metabolismo da glicose, etc. No entanto, uma pessoa não é apenas uma coleção de diferentes tipos de células, cada uma das quais toma decisões independentes. Um organismo é um sistema que possui um elo regulatório central para manter a homeostase da energia e da glicose. Este regulador principal é o hipotálamo.
O hipotálamo está localizado no diencéfalo e é ele que fornece a interconexão dos sistemas reguladores nervoso e humoral. Os neurônios hipotalâmicos percebem, processam e respondem a sinais do tecido adiposo (leptina), pâncreas (insulina) e outros estímulos hormonais (grelina, colecistocinina, polipeptídeo pancreático, etc.). O hipotálamo controla a atividade do sistema endócrino humano devido ao fato de seus neurônios serem capazes de secretar transmissores neuroendócrinos que estimulam ou inibem a produção de hormônios pela glândula pituitária. Em outras palavras, o hipotálamo, cuja massa não excede 5% do cérebro, é o centro de regulação das funções endócrinas e manutenção da homeostase de todo o organismo.
Até Dilman (Dilman V. M, "Grande relógio biológico") apontou para o papel principal do hipotálamo no desenvolvimento sistemático da disfunção metabólica, levando à obesidade, diabetes, doenças cardiovasculares, oncológicas e envelhecimento. De acordo com a teoria da hiperadaptose formada por Dilman, a sensibilidade dos receptores hipotalâmicos aos sinais provenientes dos tecidos do corpo (leptina, insulina, etc.) diminui gradualmente sistematicamente com a idade. Para provocar sua "resposta", são necessários mais e mais hormônios - mais insulina, mais leptina. Desenvolve resistência à insulina e leptina, doenças metabólicas que levam ao envelhecimento e à morte.
Dependendo das funções desempenhadas, grupos de neurônios são combinados nos núcleos do hipotálamo. Um deles - o núcleo arqueado (arqueado) é um regulador chave do comportamento alimentar e do metabolismo. Neuropeptídeos orexigênicos (estimulam o apetite) e anorexigênicos (suprimem o apetite), correspondentes aos neurônios AgRP e POMC, respectivamente, podem se formar nele. Os sinais periféricos (insulina, grelina, leptina, etc.) afetam a expressão de peptídeos que estimulam ou suprimem o apetite, o que garante a coerência da regulação central (Fig. 5).
 Fig. 5. Controle hipotalâmico do metabolismo energético. O cérebro integra sinais metabólicos (leptina, insulina, grelina, PYY3-36) de tecidos periféricos, como pâncreas, tecido adiposo e estômago. No cérebro, redes neurais especializadas coordenam mudanças adaptativas na absorção e consumo de alimentos [em 5].
Fig. 5. Controle hipotalâmico do metabolismo energético. O cérebro integra sinais metabólicos (leptina, insulina, grelina, PYY3-36) de tecidos periféricos, como pâncreas, tecido adiposo e estômago. No cérebro, redes neurais especializadas coordenam mudanças adaptativas na absorção e consumo de alimentos [em 5].Então, quem e como regula a sensibilidade dos neurônios hipotalâmicos?
Um estudo da dinâmica das mitocôndrias nos tecidos cerebrais mostrou que a dinâmica das mitocôndrias desempenha um papel significativo na capacidade dos neurônios hipotalâmicos de controlar os níveis de glicose e a homeostase energética no corpo [6,7,8].
Nos neurônios AgRP (neurônios AgRP promotores da fome), que estimulam o apetite e regulam o ganho de peso, a fome leva à divisão mitocondrial e a alimentação com alto teor de gordura leva à fusão. Ou seja, a resposta das mitocôndrias é diferente daquela na maioria das outras células.
A fusão do MX nesses neurônios regula a atividade elétrica em resposta a uma dieta rica em gordura, estimulando a produção de um peptídeo orexigênico (peptídeo AgRP), necessário para o ganho de peso e a deposição de gordura com excesso de nutrientes. As deleções de Mfn1 e Mfn2 nesses neurônios resultaram em menor ganho de peso em ratos devido a níveis mais baixos de leptina em circulação.
Os neurônios POMC (suprimir o apetite) têm a função oposta, e a dinâmica das mitocôndrias em resposta à ingestão de nutrientes é diferente. Uma diminuição na expressão de mitofusinas nesses neurônios leva a uma interrupção na conexão das mitocôndrias com o EPS e, como resultado, hiperfagia, resistência à leptina e obesidade. Ao mesmo tempo, o consumo de alimentos aumentou e o consumo de energia diminuiu.
Assim, a resposta do corpo a uma dieta rica em gordura depende de padrões de dinâmica mitocondrial em neurônios hipotalâmicos. A remodelação das mitocôndrias nos neurônios fornece sua resposta à ingestão de nutrientes, estimula a produção de neuropeptídeos que estimulam ou suprimem o apetite, afetando o metabolismo no nível do corpo (Fig. 6).
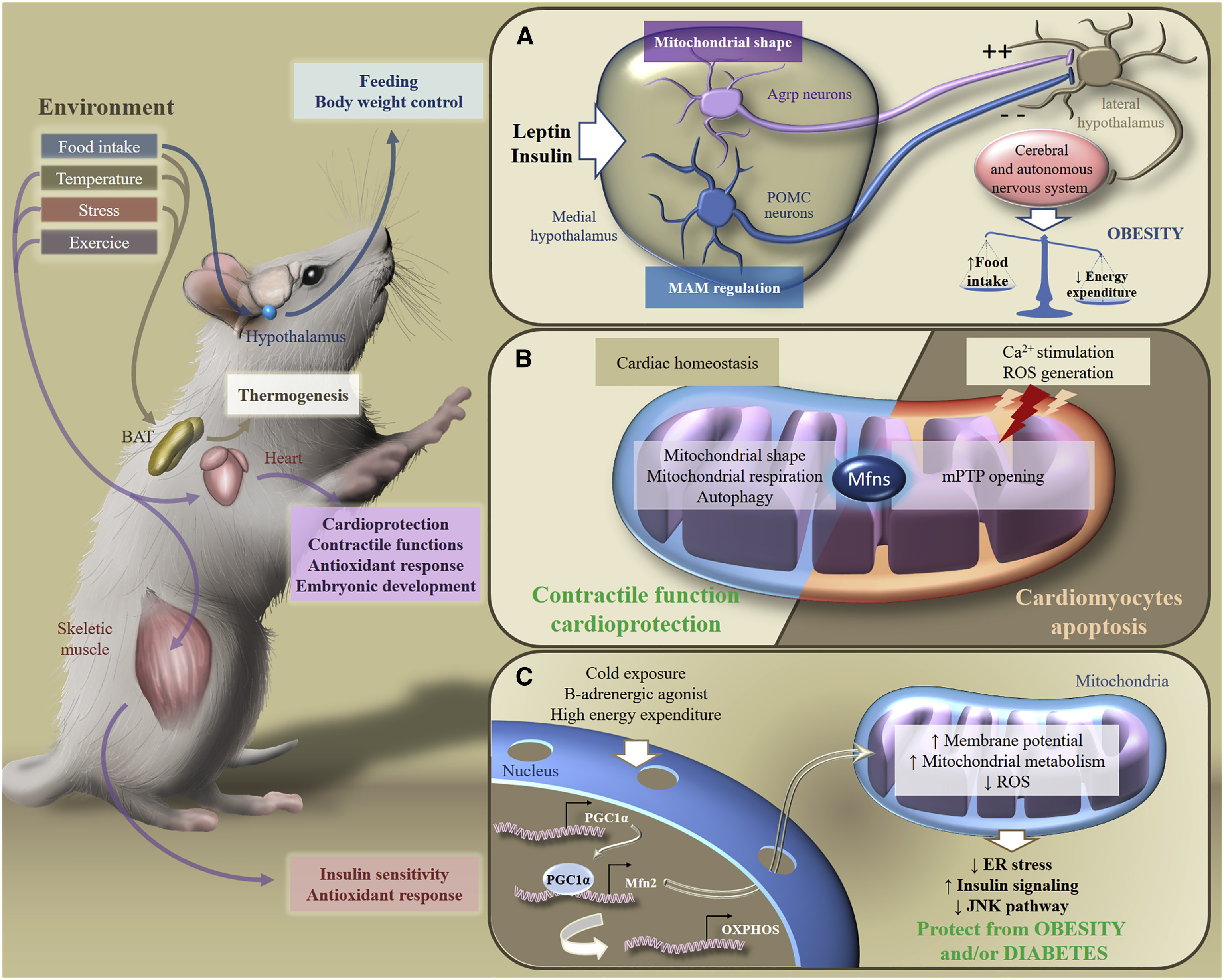 Fig. 6. Adaptação metabólica a estímulos ambientais [de 2]
Fig. 6. Adaptação metabólica a estímulos ambientais [de 2]Em resposta a estímulos exógenos, os Mfns estão envolvidos na transdução da sinalização metabólica em vários órgãos, o que garante a manutenção da homeostase energética em todo o corpo. Em particular, em resposta à ingestão de alimentos, mudanças de temperatura, estresse ou exercício, tecido adiposo marrom, cérebro, coração ou músculo esquelético adapta seu metabolismo para controlar nutrição, peso corporal, função contrátil, resposta antioxidante ou sensibilidade à insulina.
Como influenciar a dinâmica das mitocôndrias?
1. Nutrição e exercícioCiclos de nutrição O excesso de alimentos e uma dieta hiperlipídica (HFD) inibem a fusão mitocondrial nas células (o mecanismo é diferente em alguns neurônios do cérebro). Um ciclo incompleto de fissão-fusão mitocondrial interrompe os processos de autofagia → aumenta a heterogeneidade mitocondrial intracelular → não há remoção seletiva de mitocôndrias → mitocôndrias com disfunção acumulada.
A restrição calórica (ciclo de alimentação / jejum) estimula a adaptação bioenergética, fornecendo mecanismos de qualidade mitocondrial.
2. Membranas saudáveis: ácido esteárico, cardiolipina, ácido fosfatídicoTodos os processos-chave dependem da "saúde" das membranas mitocondriais - autofagia, mitofagia, apoptose, a relação das mitocôndrias com o retículo endoplasmático e a dinâmica das mitocôndrias. Membranas de organelas celulares são compostas de lipídios e proteínas. A remodelação dessas membranas é controlada por interações entre lipídios e proteínas específicos.
Os ácidos graxos saturados incluem palmítico (C16) e esteárico (C18). Foi demonstrado que o uso de ácido esteárico (C18: 0) estimula o processo de fusão mitocondrial. Sua ação está associada a um efeito sobre as mitofusinas. Nos ratos, os suplementos alimentares de ácido esteárico podem restaurar parcialmente a disfunção mitocondrial causada por mutações nos genes Pink1 ou parkin. Em neutrófilos de pessoas que seguem uma dieta baixa em C18: 0 por 2 dias, as mitocôndrias estão em um estado fragmentado (50% das células tinham MX fragmentado, 10% conectado a MX). O uso de ácido esteárico levou-os a fundir mitocôndrias após 3 horas [8]. Assim, o ácido estérico é importante para manter a dinâmica mitocondrial. A maioria do ácido esteárico é encontrada nos grãos de cacau (31-34%).
Os fosfolipídios são os principais componentes das membranas organelares. Eles também regulam a dinâmica das mitocôndrias e seu efeito é diferente [9].
A cardiolipina (CL) estimula a divisão mitocondrial e a fusão das membranas internas.
A cardiolipina é necessária para a operação do complexo IV (citrocromo C oxidase) da cadeia de transporte de elétrons. A cardiolipina está localizada quase exclusivamente na membrana interna das mitocôndrias. Com a idade, há uma diminuição na quantidade de cardiolipina. Existe uma teoria de que a perda da função da cardiolipina está associada à substituição de ácidos graxos saturados em sua molécula por ácidos graxos poliinsaturados. Para resolver esse problema, é necessário introduzir gorduras saturadas ricas em ácido graxo esteárico na dieta.
Para aumentar a eficiência da entrega de ácidos graxos saturados à membrana, os transportadores podem ser usados. Por exemplo, o uso de fosfatidilcolina saturada (dipalmitofosfatidilcolina e diseroilfosfatidilcolina), que poderia potencialmente fornecer AGs saturados diretamente à cardiolipina [10]. A colina, como transportadora, passa facilmente pelo citosol e entra nas mitocôndrias.
O ácido fosfatídico (AR) inibe a divisão mitocondrial e estimula a fusão de membranas externas (Fig. 7).
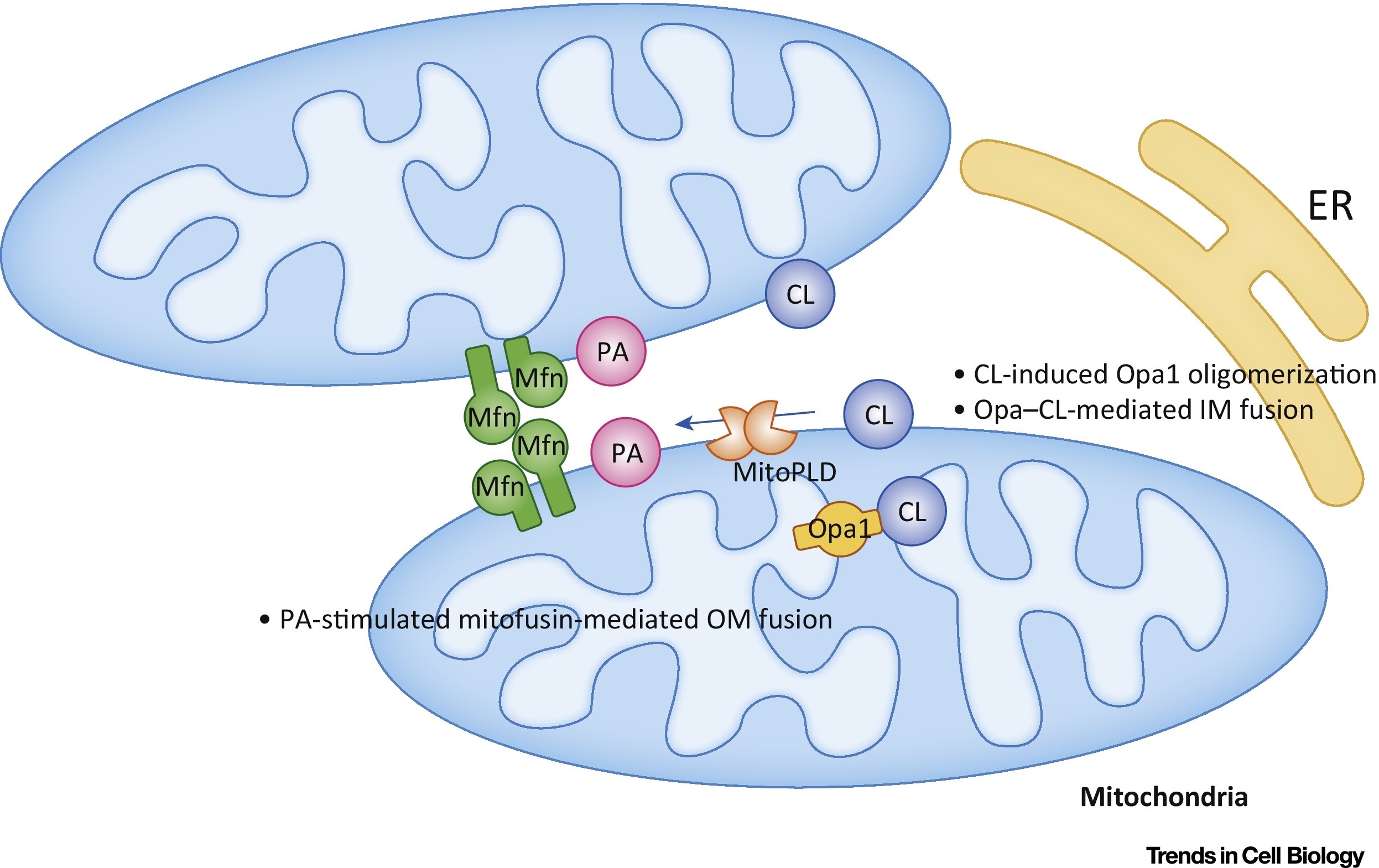 Fig. 7 Regulação da fusão mitocondrial com ácido fosfatídico (PA) e cardiolipina (CL) [em 9].
Fig. 7 Regulação da fusão mitocondrial com ácido fosfatídico (PA) e cardiolipina (CL) [em 9].Na membrana externa (OM), a AR estimula a fusão mediada por mitofusina (Mfn). Na membrana interna (IM), o CL estimula a fusão mediada por Opa1. Abreviações: ER - retículo endoplasmático; MitoPLD, - fosfolipase D. localizada na mitocôndria
3. Regulação da expressão de mitofusinas (proteínas responsáveis pela dinâmica das mitocôndrias)Tudo o que falamos acima (restrição calórica, ácido esteárico, fosfolipídios) atua afetando a expressão das mitofusinas.Além disso, existem vários medicamentos que podem afetar indiretamente a dinâmica das mitocôndrias. Isso inclui o uso de metformina.O mais interessante é o uso de substâncias que podem afetar diretamente a expressão de mitofusinas. Um dos medicamentos em potencial chamado leflunomida (leflunomida), aprovado pela FDA [5,11]. É um indutor da expressão de Mfn1 e Mfn2 e foi registrado como uma droga para o tratamento da artrite reumatóide.Terapia gênica mitocondrial
A dinâmica mitocondrial prejudicada pode estar associada à expressão prejudicada de proteínas responsáveis pela fusão e divisão mitocondrial. Além disso, a disfunção dessas proteínas pode estar associada (e isso ocorre com mais frequência) às suas mutações. Existem duas abordagens para a consideração das interações causa-efeito da disfunção das mitocôndrias.Anteriormente, acreditava-se que o estilo de vida, incluindo comer demais, leva à formação de radicais livres, estresse oxidativo, mutações do genoma mitocondrial e, consequentemente, disfunção das mitocôndrias. No entanto, recentemente, existem evidências convincentes de que mutações no DNA mitocondrial são inevitáveis, todas têm mutações pontuais no DNA heteroplásmico e estão associadas a erros de replicação, e não a danos oxidativos, aos quais o DNA mitocondrial é bastante estável [12]. Já na fase do óvulo fertilizado, algumas de nossas mitocôndrias carregam mutações. Com o tempo, eles se dividem, há mais mitocôndrias mutantes, não conseguem desempenhar sua função normalmente.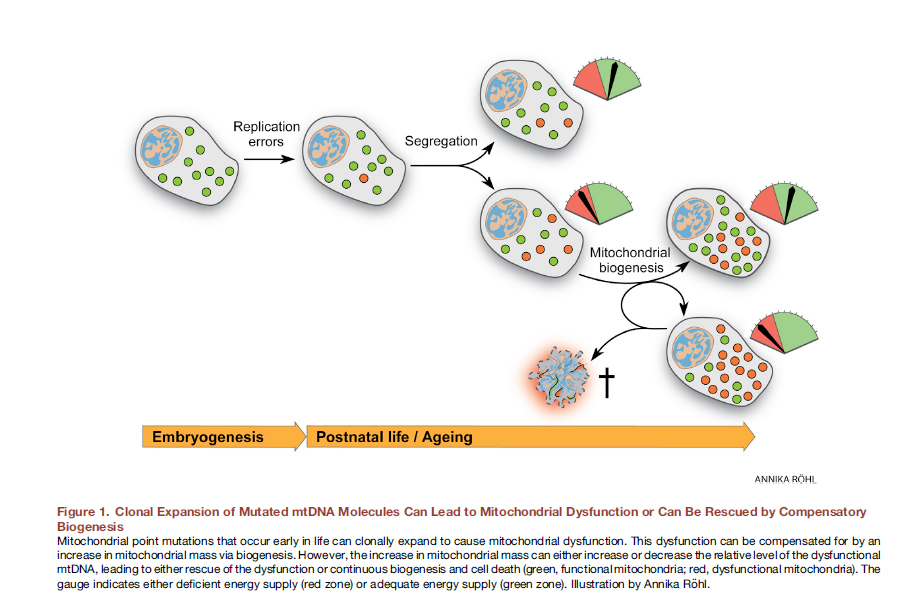 Fig. 8 A expansão clonal de moléculas de mtDNA mutadas pode levar à disfunção mitocondrial ou pode ser "salva" pela biogênese compensatória [em 12].Aqui, in vivo, a edição do genoma mitocondrial pode ser muito útil. Foi demonstrado que, para mutações pontuais de DNA heteroplasmático em camundongos, já foi alcançado um sucesso significativo com nucleases direcionadas de dedo de zinco (mtZFN) com entrega usando um vetor adenoviral [13].
Fig. 8 A expansão clonal de moléculas de mtDNA mutadas pode levar à disfunção mitocondrial ou pode ser "salva" pela biogênese compensatória [em 12].Aqui, in vivo, a edição do genoma mitocondrial pode ser muito útil. Foi demonstrado que, para mutações pontuais de DNA heteroplasmático em camundongos, já foi alcançado um sucesso significativo com nucleases direcionadas de dedo de zinco (mtZFN) com entrega usando um vetor adenoviral [13].Transferência mitocondrial
Outro método promissor para eliminar a disfunção mitocondrial é o transplante mitocondrial. A essência dessa abordagem é "substituir" as mitocôndrias danificadas por mitocôndrias exógenas saudáveis. Essa abordagem foi usada clinicamente pela primeira vez em crianças com isquemia miocárdica. Mitocôndrias isoladas autólogas foram usadas para transplante, as quais foram isoladas com o músculo reto abdominal (foi realizada biópsia e, em seguida, a preparação foi preparada) e depois administradas por injeção direta [14]. Várias abordagens para a introdução de mitocôndrias estão sendo desenvolvidas: injeção direta de mitocôndrias isoladas (injeção local) e injeção sistêmica na corrente sanguínea, quando as próprias mitocôndrias “buscam” por qual célula ir. Grupos de pesquisadores estão estudando a possibilidade de transplante mitocondrial na doença de Parkinson, isquemia hepática, acidente vascular cerebral, doenças mitocondriais [15].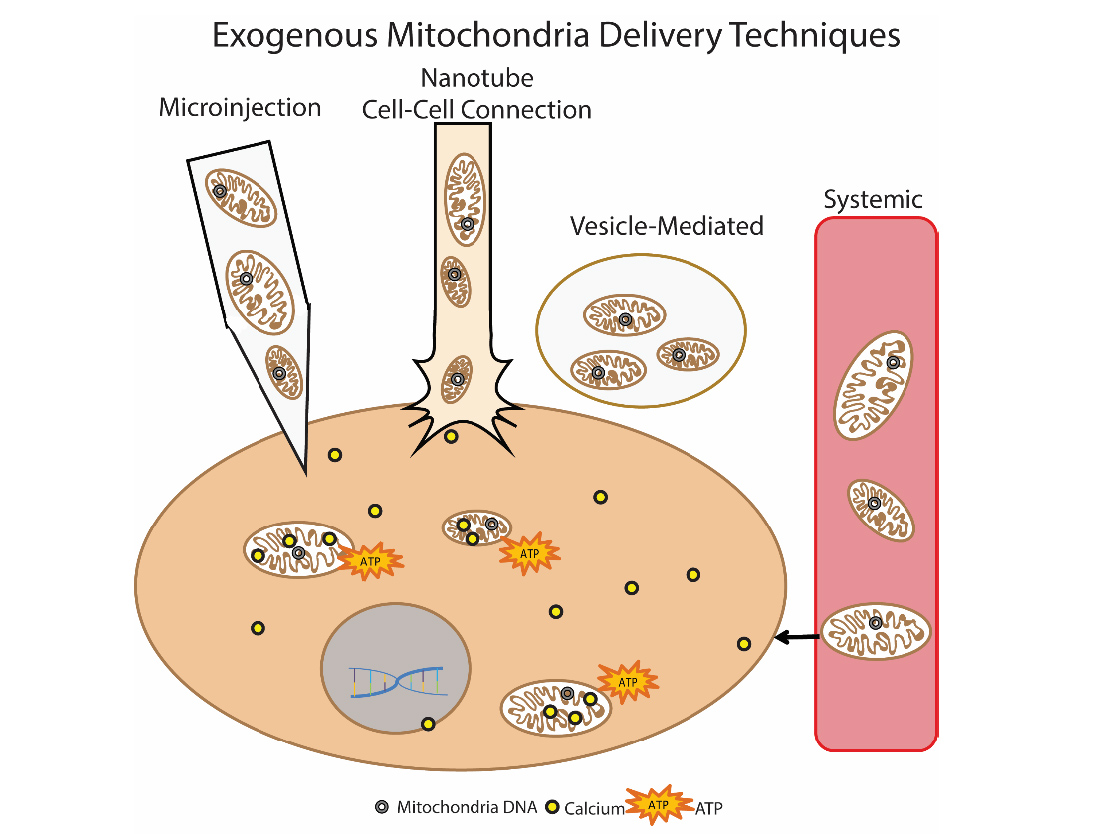 Fig. 9 Métodos para a entrega de mitocôndrias exógenas à célulaAutor Olga Borisova
Fig. 9 Métodos para a entrega de mitocôndrias exógenas à célulaAutor Olga BorisovaLiteratura1. Kauppila, Timo ES, Johanna HK Kauppila, and Nils-Göran Larsson. «Mammalian mitochondria and aging: an update.» Cell metabolism 25.1 (2017): 57-71.
www.sciencedirect.com/science/article/pii/S15504131163050222. Schrepfer, Emilie, and Luca Scorrano. «Mitofusins, from mitochondria to metabolism.» Molecular cell 61.5 (2016): 683-694.
www.sciencedirect.com/science/article/pii/S1097276516001337#fig13. Marc Liesa, Orian Shirihai “Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure” Cell methabolism (2013): 491-506
www.sciencedirect.com/science/article/pii/S1550413113001046#fig34. Ramos, Eduardo Silva, Nils-Göran Larsson, and Arnaud Mourier. «Bioenergetic roles of mitochondrial fusion.» Biochimica et Biophysica Acta (BBA)-Bioenergetics 1857.8 (2016): 1277-1283.
www.sciencedirect.com/science/article/pii/S00052728163008585. Cunarro, Juan, et al. «Hypothalamic mitochondrial dysfunction as a target in obesity and metabolic disease.» Frontiers in endocrinology 9 (2018): 283.
www.frontiersin.org/articles/10.3389/fendo.2018.00283/full6. Marcelo O.Dietrich et al. «Mitochondrial Dynamics Controlled by Mitofusins Regulate Agrp Neuronal Activity and Diet-Induced Obesity”.
www.sciencedirect.com/science/article/pii/S0092867413010957#figs27. Steculorum, Sophie M., and Jens C. Brüning. „Sweet mitochondrial dynamics in VMH neurons.“ Cell metabolism 23.4 (2016): 577-579.
www.sciencedirect.com/science/article/pii/S15504131163011768. Senyilmaz-Tiebe, Deniz, et al. „Dietary stearic acid regulates mitochondria in vivo in humans.“ Nature communications 9.1 (2018): 3129.
www.nature.com/articles/s41467-018-05614-69. Kameoka, Shoichiro, et al. „Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics.“ Trends in cell biology (2017).
www.sciencedirect.com/science/article/pii/S096289241730158710.
raypeatforum.com/community/threads/mitolipin-liquid-saturated-phosphatidylcholine-pc-mix.1039811. Miret-Casals, Laia, et al. „Identification of new activators of mitochondrial fusion reveals a link between mitochondrial morphology and pyrimidine metabolism.“ Cell chemical biology25.3 (2018): 268-278.
12. Kauppila, Timo ES, Johanna HK Kauppila, and Nils-Göran Larsson. „Mammalian mitochondria and aging: an update.“ Cell metabolism 25.1 (2017): 57-71.
13. Gammage et al. “Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo” Nature medicine, 2017
www.nature.com/articles/s41591-018-0165-914. Emani, Sitaram M., et al. „Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury.“ The Journal of thoracic and cardiovascular surgery 154.1 (2017): 286-289.
www.jtcvs.org/article/S0022-5223 (17)30258-1/fulltext
15. McCully, James D., et al. „Mitochondrial transplantation: From animal models to clinical use in humans.“ Mitochondrion 34 (2017): 127-134.
www.sciencedirect.com/science/article/pii/S1567724917300053